图解blast验证引物教程1
DNA序列比对同源性分析图解BLAST
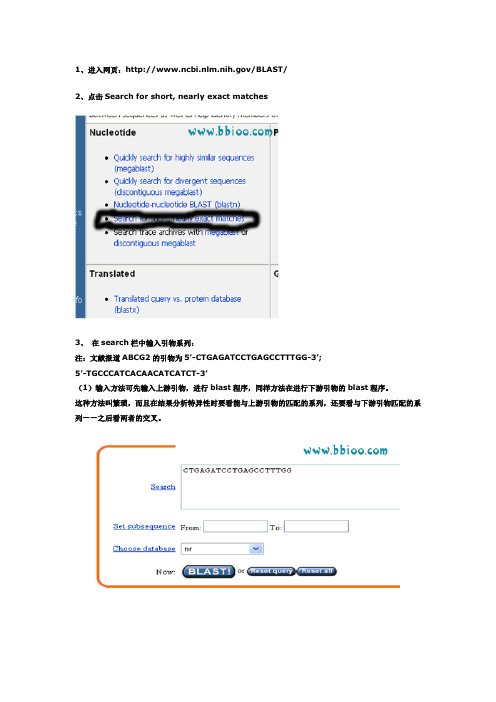
1、进入网页:/BLAST/2、点击Search for short, nearly exact matches3、在search栏中输入引物系列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物B、输入上游引物回车输入下游引物4、在options for advanced blasting中:select from 栏通过菜单选择Homo sapiensExpect后面的数字改为105、在format中:select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 106、点击网页中最下面的“BLAST!”7、出现新的网页,点击Format!8、等待若干秒之后,出现results of BLAST的网页。
该网页用三种形式来显示blast的结果。
(1)图形格式:图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:从左到右分别为:A、数据库系列的身份证:点击之后可以获得该序列的信息B、系列的简单描述C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
按照得分的高低由大到小排列。
得分的计算公式=匹配的碱基×2+0.1。
DNA序列比对同源性分析图解BLAST
1、进入网页:/BLAST/2、点击Search for short, nearly exact matches3、在search栏中输入引物系列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物B、输入上游引物回车输入下游引物4、在options for advanced blasting中:select from 栏通过菜单选择Homo sapiensExpect后面的数字改为105、在format中:select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 106、点击网页中最下面的“BLAST!”7、出现新的网页,点击Format!8、等待若干秒之后,出现results of BLAST的网页。
该网页用三种形式来显示blast的结果。
(1)图形格式:图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:从左到右分别为:A、数据库系列的身份证:点击之后可以获得该序列的信息B、系列的简单描述C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
按照得分的高低由大到小排列。
得分的计算公式=匹配的碱基×2+0.1。
基于NCBI-primer blast的引物设计及验证

基于NCBI-primer blast的引物
设计及验证
流程
01 02引物的设计(已知模板设计引物)
引物的验证(已知引物查找模板/验证参数)
1-primer blast中引物的设计——模板
可以放序列:
如
也可以放NM号:
如NM_001289746.2
预期引物Tm 值:最适60,无特殊要求无需更改
预期产物大小:如80-150bp
数据库来源:根据模板设置,模板是
cDNA 就选mRNA
跨外显子设置:
RT-qPCR 定量引物为避免gDNA 干扰需设置跨外显子
物种种属设置:根据模板的种属设置
1-primer blast 中引物的设计——生成引物
生成引物:点击生成10对引物,可进行选择
最大扩增子设置:可不做更改,引物设计时前面已设置过产物大小
2-primer blast中已知引物的验证——只需放入引物序列
引物序列:
待验证引物序列放
于此处,注意引物
方向
2-primer blast中已知引物的验证——设置来源和种属
待验证引物来源数据库:
即是基于cDNA(mRNA反转)
的扩增引物,还是基于
gDNA的扩增引物;如定量
则肯定选mRNA
待验证引物来源种属:
即该引物扩增的模板的种属
注:如这两项未知,可先不做选择进行验证;如得不到完全匹配的结果,再进
行更改。
2-primer blast中已知引物的验证——进行验证
验证引物:
点击即可对上述输
入的引物进行验证,
可看到包括Tm值,
扩增子大小等引物
参数。
blast验证引物

程序的选择
点击此按钮, 进入结果页面
等待若干秒之后,出现results of BLAST的网页。 该网页用三种形式来显示blast的结果。
(1)图形格式 通过点击相应的bar 可以得到匹配情况的 详细信息。
(2)结果信息概要:
A B C D E
从左到右分别为: A、数据库系列的身份证:点击之后可以获得该序列的信息 B、系列的简单描述 C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。按照得分 的高低由大到小排列。得分的计算公式=匹配的碱基×2+0.1。举例:如果有20个碱 基匹配,则其得分为40.1。 D、E值:代表被比对的两个序列不相关的可能性。E值最低的最有意义,也就 是说序列的相似性最大。设定的E值是我们限定的上限,E值太高的就不显示了 E、最后一栏有的有UEG的字样,其中: U代表:Unigene数据库 E代表:GEO profiles数据库 G代表:Gene数 据库
blast验证引物
一、概述
BLAST是Basic Local Alignmen类似性检索工具。它采用统计学记分系统,能将真 正配对的序列同随机产生的干扰序列区别开来;同时 采用启发式算法系统,即采用的是局部对准算法 (Local Alignment Algorithm),而不是全序列对准算 法(Global Alignment Algorithm)。
特殊blast
先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻 译成蛋白质序列,然后再将两种翻译结果从蛋白质水平进行查询。
ATGTTGGGGAAATGCTTGACC
引物序 列输入 窗口 数据库的选择 在此,我们选择 第三个数据库 输入方法可先输入上游引物,进行blast程序,同样 方法在进行下游引物的blast程序。在结果分析特异 性时要看能与上游引物的匹配的系列,还要看与下 游引物匹配的系列——之后看两者的交叉。 显示结 果在新 的窗口
如何用Primer-Blast设计和验证引物

如何⽤Primer-Blast设计和验证引物——⽇读⼀帖,解螺旋⼤V团队伴你科研路【科研热点】让你时间⽐别⼈花的少、知道的⽐他早!【基⾦专栏】国⾃然等各项基⾦独到经验见解【SCI 专栏】从开始到接收全程tips【实验技能】这么棒快告诉你⽼板! 关注我们,为您的科研路提速 今天⽼谈给⼤家推荐NCBI的⼀款在线⼯具Primer-BLAST,⽤于PCR的特异性引物设计和特异性检验。
推荐的指数:5颗星。
理由:操作简单,使⽤⽅便,不需要安装程序,⽽且和NCBI数据库已⽐对,不⽤担⼼特异性问题。
理由:⼀、Primer-BLAST介绍 Primer-BLAST可以直接从Blast主页(/)找到,或是直接⽤下⾯的链接进⼊:/tools/primer-blast/,这个⼯具整合了⽬前流⾏的Primer3软件,再加上NCBI的Blast进⾏引物特异性的验证。
Primer-BLAST免除了⽤另⼀个站点或⼯具设计引物的步骤,设计好的引物程序直接⽤Blast进⾏引物特异性验证。
更强⼤的是Primer-BLAST能设计出只扩增某⼀特定剪接变异体基因的引物。
Primer-BLAST有许多改进的功能,⽐单个的⽤Primer3和NCBI BLAST更加准确。
⼆、Primer-BLAST的输⼊ Primer-BLAST界⾯包括了Primer3和BLAST的功能。
提交的界⾯主要包括4部分:PCR Template(模板区), Primer Parameters(引物区), Exon/intron selection(外显⼦内含⼦设置)和specificity check(特异性验证区)。
(1)模板(Template) 在“PCR Template”下⾯的⽂本框,输⼊⽬标模板的序列,FASTA格式或直接⽤Accession Number。
如果你在这⾥输⼊了序列,是⽤于引物的设计。
Primer-BLAST就会根据你输⼊的序列设计特异性引物,并且在⽬标数据库(在specificity check区选择)是唯⼀的。
如何用Primer-Blast设计和验证引物

如何用Primer-Blast设计和验证引物——日读一帖,解螺旋大V团队伴你科研路【科研热点】让你时间比别人花的少、知道的比他早!【基金专栏】国自然等各项基金独到经验见解【SCI 专栏】从开始到接收全程tips【实验技能】这么棒快告诉你老板!关注我们,为您的科研路提速今天老谈给大家推荐NCBI的一款在线工具Primer-BLAST,用于PCR的特异性引物设计和特异性检验。
推荐的指数:5颗星。
理由:操作简单,使用方便,不需要安装程序,而且和NCBI数据库已比对,不用担心特异性问题。
一、Primer-BLAST介绍Primer-BLAST可以直接从Blast主页(/)找到,或是直接用下面的链接进入:/tools/primer-blast/,这个工具整合了目前流行的Primer3软件,再加上NCBI的Blast进行引物特异性的验证。
Primer-BLAST免除了用另一个站点或工具设计引物的步骤,设计好的引物程序直接用Blast进行引物特异性验证。
更强大的是Primer-BLAST能设计出只扩增某一特定剪接变异体基因的引物。
Primer-BLAST有许多改进的功能,比单个的用Primer3和NCBI BLAST更加准确。
二、Primer-BLAST的输入Primer-BLAST界面包括了Primer3和BLAST的功能。
提交的界面主要包括4部分:PCR Template(模板区), Primer Parameters (引物区), Exon/intron selection(外显子内含子设置)和specificity check(特异性验证区)。
(1)模板(Template)在“PCR Template”下面的文本框,输入目标模板的序列,FASTA格式或直接用Accession Number。
如果你在这里输入了序列,是用于引物的设计。
Primer-BLAST就会根据你输入的序列设计特异性引物,并且在目标数据库(在specificity check区选择)是唯一的。
Primer-BLASTNCBI的引物设计和特异性检验工具

Primer-BLAST:NCBI的引物设计和特异性检验工具Primer-Blast介绍Primer-BLAST,在线设计用于聚合酶链反应(PCR)的特异性寡核苷酸引物。
Primer-BLAST可以直接从Blast主页()找到,或是直接用下面的链接进入:这个工具整合了目前流行的Primer3软件,再加上NCBI的Blast进行引物特异性的验证。
Primer-BLAST免除了用另一个站点或工具设计引物的步骤,设计好的引物程序直接用Blast进行引物特异性验证。
并且,Primer-BLAST能设计出只扩增某一特定剪接变异体基因的引物–an important feature for PCR protocols measuring tissue specific expression(注:没办法准确的翻译,只好作罢,汗!)。
Primer-BLAST有许多改进的功能,这样在选择引物方面比单个的用Primer3和NCBI BLAST更加准确。
Primer-BLAST的输入Primer-BLAST界面包括了Primer3和BLAST的功能。
提交的界面主要包括三个部分:target template(模板区), the primers(引物区), 和specificity check(特异性验证区)。
跟其它的BLAST一样,点击底部的“Advanced parameters”有更多的参数设置。
模板(Template)在“PCR Template”下面的文本框,输入目标模板的序列,FASTA格式或直接用Accession Number。
如果你在这里输入了序列,是用于引物的设计。
Primer-BLAST 就会根据你输入的序列设计特异性引物,并且在目标数据库(在specificity check区选择)是唯一的。
引物(Primers)如果你已经设计好了引物,要拿来验证引物的好坏。
可以在Primer Parameters 区填入你的一条或一对引物。
如何运用BLAST进行序列比对、检验引物特异性之欧阳数创编

序列比对,绝大多数战友都会想到BLAST,但BLAST的使用确实又是一个很大的难题,因为他的功能比较强悍,里面涉及到的知识比较多,而且比对结束后输出的结果参数(指标)又很多。
如果把BLAST 的使用详细的都讲出来,我想我发帖发到明天也发不完,更何况我自己也不是完全懂得BLAST的使用。
所以我在这里也就“画龙点睛”——以比对核酸序列为例来给大家介绍一下BLAST的使用,也算是BLAST的入门课程吧。
请看帖的战友好好体会,如果你用心看,在看帖完毕之后BLAST的基本使用(包括其他序列的比对)应该没有问题了。
一、打开BLAST页面,http://www.ncbi.nlm.nih.go/BLAST/ 打开后如图所示:(缩略图,点击图片链接看原图)对上面这个页面进行一下必要的介绍:BLAST的这个页面主体部分(左面)包括了三部分:BLAST Assembled Genomes、Basic BLAST、Specialized BLAST。
相信大家可以看懂这三个短语的意思,我就不多说了;我要说的是,可以认为这是三种序列比对的方法,或者说是BLAST的三条途径。
第一部分BLAST Assembled Genomes就是让你选择你要比对的物种,点击相应物种之后即可进入比对页面。
第二部分Basic BLAST包含了5个常用的BLAST,每一个都附有简短的介绍。
第三部分Specialized BLAST是一些特殊目的的BLAST,如IgBLAST、SNP等等,这个时候你就需要在Specialized BLAST部分做出适当的选择了。
总之,这是一个导航页面,它的目的是让你根据自己的比对目的选择相应的BLAST途径。
下面以最基本的核酸序列比对来谈一下BLAST 的使用,期间我也会含沙射影的说一下其他序列比对的方法。
二、点击Basic BLAST部分的nucleotide blast链接到一个新的页面。
打开后如图所示:screen.width-333)this.width=screen.width-333" width=640 height=462title="Click to iew full2.JPG (849 X 613)" border=0 align=absmiddle> 介绍一下上述页面:Enter Query Sequence部分是让我们输入序列的,你可以直接把序列粘贴进去,也可以上传序列,还可以选择你要比对的序列的范围(留空就代表要比对你要输入的整个序列)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
图解blast 验证引物教程
——以文献报道的人类的ABCG2的引物为例
1、 进入网页:/BLAST/
2、 点击Basic BLAST 中的nucleotide blast 选项
3、 完成2操作后就进入了Basic Local Alignment Search Tool 界面 (1)在Enter Query Sequence 栏中输入引物序列:
注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;
5’-TGCCCATCACAACATCATCT-3’
简便的做法是同时输入上下游引物。
输入上下游引物系列都从5’— 3’。
输入上游引物后,加上≥20个字母n ,再输入下游引物,如下图:
生 物 秀
(2)在Choose Search Set 栏中:
Database 根据预操作基因的种属定了,本引物可选Human genomic + transcript
或Others (nr etc.)。
本人倾向于选后者,觉得此库信息更多。
如下图:
(3)在Program Selection 中:选择Somewhat similar sequences (blastn)项,如下图:
(4)在此界面最下面:如下图
生物秀-专心做生物
w w w .b b i o o .c o m
Show results in a new window 项是显示界面的形式,可选可不选,在此我们选上了。
关键要点击Algorithm parameters 参数设置,进入参数设置界面。
4. 参数设置:
(1)在General Parameters 中:Expect thresshold 期望阈值须改为1000,大于1000也可以;在Word size 的下拉框将数字改为7。
如下图:
(2)Scoring Parameters 无须修改
(3)Filters and Masking 中,一般来说也没有必要改
5.点击最下面一栏的BLAST 按钮,如图:
6.点击BLAST 按钮后,跳转出现如下界面:
7. 等待若干秒之后,自动跳转出现显示BLAST 结果的网页。
该网页用三种形式来显示blast 的结果。
生物秀-专心做生物
w w w .b b i o o .c o m
(1)图形格式:
图中①代表这些序列与引物匹配的得分值小于40分
图中②代表这些序列与引物匹配的得分值位于40~50分
图中③代表这些序列与引物匹配的得分值位于80~120分,
分值越高,特异性越好。
线段代表上或下游引物,其颜色和上面对照后就可得出该条引物的分值。
图中两线段间有连线的代表这些序列与上游引物匹配(Strand=Plus/Plus)、并与下游引物互补(Strand=Plus/Minus),理论上可以扩增出基因片断。
没有连线的,表示单条引物与该基因一致。
点击线段,就能跳转到该基因的结果信息概要。
(2)结果信息概要:
Accession 、数据库系列的身份证:点击之后可以获得该序列的信息 Desscription 、系列的简单描述
Total score 高的就是有两线段间有连线的,如图划线的。
E value :代表被比对的两个序列不相关的可能性。
【The E value decreases exponentially as the Score (S) that is assigned to a match between two sequences increases 】。
E 值最低的最有意义,也就是说序列的相似性最大。
设定的E 值是我们限定的上限,E 值太高的就不显示了
E 、最后一栏有的有UEG 的字样,其中: U 代表:Unigene 数据库
E 代表:GEO profiles 数据库 G 代表:Gene 数据库
(3)结果详细信息:
生物秀-专心做生物
w w w .b b i o o .c o m
划线上代表上游引物与该序列的正链【Plus/Plus 】的匹配情况: 划线下代表下游引物与该序列的负链【Plus/Minus 】的匹配情况: 共有20个碱基匹配,得分40.1分【20×2+0.1=40.1】,E 值为0.077。
为什么与上下游引物匹配的ABCG2序列有多种? A 、 为同一个基因来源的不同的mRNA 片段 B 、 为该基因的DNA 系列 C 、 为同一个基因来源的不同的cDNA 克隆片段。
结果判断:
①验证文献报道的引物是否正确:如果你可以在所显示的结果中找出你的目的基因,一般说明你的引物正确性没问题。
如果你blast 后没有发现你的目的基因,或者分值很低,该引物就可能不适合用
②检测该对引物是否可与其它序列匹配,引起PCR 的非特异性扩增。
如果找到了你的目的基因名称,而且找到了一大批同物种的不同基因,(上下游引物分别搜索到相同的基因),而且分数也较高。
这时表明你的引物设计的特异性不高,极有可能在你的扩增产物中出现非特异性产物。
生物秀-专心做生物
w w w .b b i o o .c o m
生物秀-专心做生物
生物秀论坛—学术交流、资源共享与互助社区
http://
生物部落—生物医药人网络家园
http://
生物百科—生命科学第一百科
http://
生物秀知道—生命科学问题解决之道!
http://
易生物-领先的生物医药商务平台。