土壤中总氰化物检测方法
土壤中有害氰化物的检测方法

土壤中有害氰化物的检测方法作者:韩康芹张云肖冯敏英来源:《安徽农业科学》2014年第03期摘要 [目的] 为了准确检测出土壤中氰化物的含量。
[方法]《生活饮用水标准检验方法》GB/T 5750-2006异烟酸-吡唑酮分光光度法检测水中氰化物的方法已被广泛采用,但是土壤基质相当复杂,测定过程中基质干扰、固体颗粒的吸附、土样保存、蒸馏、萃取等过程均有可能导致其含量的损失,直接影响检测结果的准确性。
[结果] 质量控制中,采用平行样品检测控制其精密度,加标回收率的测定控制其准确度。
[结论]检出结果能正确地反映氰化物在土壤中的含量。
关键词土壤;氰化物;检测方法;标准溶液中图分类号 S156 文献标识码 A 文章编号 0517-6611(2014)03-00729-02土壤是陆地地表能生长绿色植物的疏松层,能为植物提供水、空气和养分。
土壤是植物生长的基地,是动物、人类赖以生存的物质基础。
因此,土壤质量的优劣直接影响人类的生存和发展。
但是,由于近些年人们不合理的开发和利用,许多污染物质通过多种渠道进入土壤,特别是随着石油化工、塑料、合成纤维、焦化等工业和农业的迅速发展,土壤污染的情况日趋严重。
氰化物属剧毒、高毒物质。
极少量的氰化物(每千克体重数毫克)就会使人畜在很短的时间内中毒死亡。
含氰化物浓度很低的水(土壤基质相当复杂。
氰化物与土壤中有机质含量有一定的相关性。
在测定过程中基质干扰、固体颗粒的吸附、土样保存、蒸馏、萃取等均有可能导致其含量的损失,直接影响检测结果的准确性。
该研究是在大量研究基础上的总结。
检出结果能正确反映出氰化物在土壤中的含量。
1 土壤样品的保存氰化物易挥发,所以采样时应用干净的棕色广口玻璃瓶(或聚乙烯瓶)采样,瓶口要密封。
样品采集后应尽快检测,如需要短期保存,应置于冰箱内4 ℃以下避光保存,可保存期为2 d。
2 检测仪器用分光光度计进行检测。
3 主要试剂3.1 一般试剂磷酸盐缓冲溶液、氯胺T水溶液(10 g/L)、吡啶—吡唑啉酮溶液、乙酸锌溶液(100g/L)、酒石酸溶液(150 g/L)、氢氧化钠溶液(10 g/L)、酚酞指示剂(10 g/L)。
土壤氰化物的测定

土壤氰化物的测定土壤中的氰化物是一种常见的有毒物质,对人体健康和环境造成严重威胁。
因此,为了保护人类和环境的健康,对土壤中的氰化物含量进行准确测定是非常重要的。
本文将介绍土壤氰化物测定的方法及其应用。
氰化物是一种非常活泼的离子,它与许多金属离子能够形成稳定的氰配合物,并在一定程度上改变土壤中金属离子的生物利用度。
由于氰化物的有毒性和易溶性,如果土壤中氰化物的含量超过一定的浓度,将对生态系统产生严重影响。
因此,准确测定土壤中的氰化物含量对于监测土壤质量、评估环境污染、指导农作物种植等方面具有重要意义。
测定土壤氰化物的方法繁多,常见的方法包括光谱法、离子选择电极法、电化学法、电感耦合等离子体发射光谱法(ICP-AES)等。
其中,离子选择电极法是一种常用且准确测定土壤中氰化物含量的方法。
这种方法依靠离子选择电极与氰化物离子之间的化学反应,通过测量电极产生的电位变化来检测氰化物的浓度。
在进行土壤氰化物测定之前,需要对土壤样品进行预处理。
首先,收集土壤样品,并根据需要进行分层采样。
然后,将土壤样品在室温下晾干,并通过过筛网将粗颗粒物去除。
接下来,将土壤样品与适量的溶剂(通常为水)混合,并在震荡器中振荡一段时间,以便溶剂充分与土壤样品接触,溶解土壤中的氰化物。
最后,通过离心将溶剂和土壤样品分离,得到土壤溶液。
接下来,使用离子选择电极或其他适当的仪器对土壤溶液中的氰化物含量进行测定。
使用离子选择电极时,将电极浸入土壤溶液中,并根据仪器的使用说明进行操作。
仪器将测量电极在土壤溶液中产生的电位变化,并根据标准曲线计算出土壤中氰化物的浓度。
除了离子选择电极法,还可以使用其他方法对土壤中的氰化物进行测定。
例如,可以使用高效液相色谱仪(HPLC)进行分析,通过色谱柱分离氰化物,并通过检测器测量氰化物的浓度。
此外,也可以使用气相色谱仪(GC)对土壤中的挥发性氰化物进行测定。
这些方法都可以根据实际需求选择合适的方法进行分析。
66 土壤 氰化物总氰化物745-2015(1)
相对湿度:%
标液名称配制浓度编来自/批号配制日期
工作曲线
分取标液体积(mL)
口浓度口含量x()
吸光度值(A-A0)
(空白值A0=)
回归方程
y=a+bx a= b=
相关系数r =
质控方式
口加标回收
加标量:
本底值:
测定值:
回收率(%):
口中间浓度点校准
标液浓度值:
测定值:
相对误差(%):
口质控标样
口总氰化物:打开冷凝水,在接收瓶中加入10ml氢氧化钠溶液作为吸收液。在加入试样后的蒸馏瓶中,依次加200ml水、3.0ml氢氧化钠溶液、2.0ml氯化亚锡溶液和10ml硫酸铜溶液摇匀,迅速加入10ml磷酸,立即盖塞。
打开电炉,由低档逐渐升高,馏出液以2ml/min~4ml/min速度进行加热蒸馏。接收瓶内试样近100ml时,停止蒸馏,用少量水冲洗馏出液导管后取出接收瓶,用水定容(V1),此为试样A。从试样A中吸取10.0ml试料A于25ml具塞比色管中。向各管中加入5.0ml磷酸二氢钾溶液,混匀,迅速加入0.3ml氯胺T溶液,立即盖塞,混匀,放置1min~2min。向各管中加入6.0ml异烟酸-巴比妥酸显色剂,加水稀释至标线,摇匀,于25℃显色15min(15℃显色25min;30℃显色10min)分光光度计在600nm波长下,用10mm比色皿,以水为参比,测定吸光度。
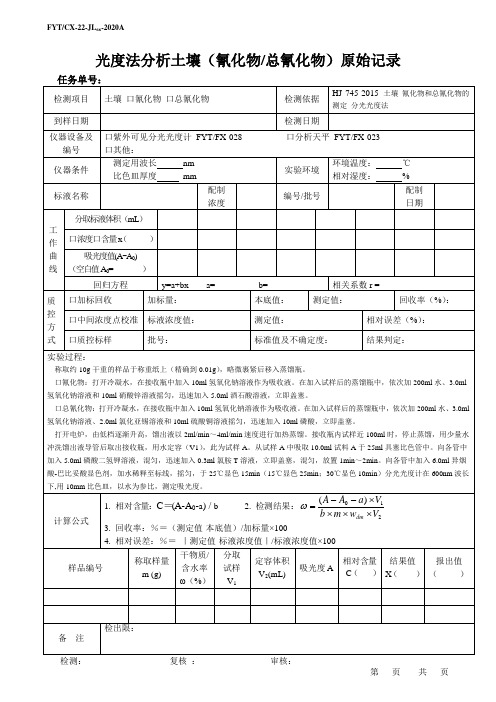
光度法分析土壤(氰化物/总氰化物)原始记录
任务单号:
检测项目
土壤口氰化物口总氰化物
检测依据
HJ 745-2015土壤氰化物和总氰化物的测定分光光度法
到样日期
检测日期
仪器设备及编号
口紫外可见分光光度计FYT/FX-028口分析天平FYT/FX-023
土壤氰化物曲线不显色

土壤氰化物曲线不显色近年来,土壤氰化物曲线逐渐成为了环境监测的重要工具。
然而,在实际使用中,我们发现有时土壤氰化物曲线不显色,这给环境监测带来了一定的困扰。
本文将从土壤氰化物的来源、检测方法、曲线显色机制等方面探讨土壤氰化物曲线不显色的原因,并提出相应的解决方案。
一、土壤氰化物的来源土壤氰化物主要来自于人类活动和自然界的生物过程。
人类活动中,农业、工业、燃煤等过程都会产生氰化物。
例如,氰化物是某些杀虫剂和杀鼠剂的主要成分,它们在使用后可能会渗入土壤中。
此外,一些工业过程中也会产生氰化物,例如金属加工、纺织品生产等。
自然界中,氰化物则主要来自于植物的生物过程。
例如,一些植物在受到损伤时会释放氰化物来抵御天敌。
二、土壤氰化物的检测方法土壤氰化物的检测方法主要有颜色反应法、电化学法和光谱法等。
其中,颜色反应法是最常用的方法之一。
该方法基于氰化物与铁离子形成化合物的化学反应,产生不同的颜色反应。
一般来说,氰化物含量越高,反应颜色越深。
这种方法简单、便宜,但其灵敏度较低,不能检测低浓度的氰化物。
电化学法则是一种高灵敏度的氰化物检测方法。
该方法通过测量氰化物与电极反应的电流或电势差来确定其浓度。
然而,该方法需要较为精密的仪器设备,成本较高。
光谱法是一种新兴的检测方法,该方法利用物质吸收或发射光线的特性来测量其浓度。
该方法具有高灵敏度、高选择性和快速分析的优点,但其设备价格昂贵,不适用于大规模的土壤氰化物监测。
三、土壤氰化物曲线不显色的原因在使用颜色反应法检测土壤氰化物时,有时会出现曲线不显色的情况。
这可能是由于以下原因所致:(1)土壤样品中含有过多的干扰物质,例如铁、铜等离子,这些离子会影响氰化物与铁离子反应的颜色变化。
(2)氰化物浓度过低,未达到颜色反应法的检测灵敏度。
(3)颜色反应试剂的配制不当,试剂浓度过低或过高都会影响反应的颜色变化。
(4)反应时间过短,颜色反应需要一定的时间才能显现出来。
四、解决方案针对以上原因,我们可以采取以下措施:(1)优化样品处理方法,去除干扰物质,提高氰化物检测的灵敏度。
土壤 氰化物和总氰化物的测定 分光光度法 方法验证(含真实案例数据)HJ 745-2015

方法确认报告方法名称:HJ HJ 745-2015土壤氰化物和总氰化物的测定项目名称:氰化物和总氰化物的测定异烟酸-巴比妥酸分光光度法编制人:______ _____校核人:_________________批准人:_________________报告日期:年月日根据本公司《质量手册》及《程序文件》的规定,对《土壤氰化物和总氰化物的测定分光光度法》HJ HJ 745-2015中异烟酸-巴比妥酸分光光度法进行了学习和练习。
本检测报告根据《分析方法检出限和定量限的评估》(GBT 27415-2013)和《化学分析方法验证确认和内部质量控制要求》(GBT 32465-2015)进行编制。
方法验证报告如下:一、验证项目的检测项目及检测方法验证项目:土壤氰化物和总氰化物的测定检测方法:异烟酸-巴比妥酸分光光度法二、验证仪器及试剂和材料情况表1 仪器设备三、标准曲线配制标准曲线硫化物含量梯度0.00μg,0.05μg,0.25μg,0.75μg,2.00μg,5.00μg,,由低浓度到高浓度依次测试,验证方法中标准曲线测定值见表3。
表3 标准曲线绘制四、方法检出限测试数据按照《分析方法检出限和定量限的评估》(GBT 27415-2013)和《化学分析方法验证确认和内部质量控制要求》(GBT 32465-2015)中方法检出限的规定方法,按照样品分析的全部步骤,对自配含0.05ug硫化物的样品平行测定7次,用标准偏差进行计算,方法检出限见表4:表4 检出限测试结果五、方法精密度测试数据三种不同浓度的硫化物样品,用于本次方法的验证。
测试结果如下:结论:根据上述表格中的数据,本方法对不同浓度的标准样品,其测试的精密度良好,满足测试要求。
六、准确度的验证对能进行加标回收的,需要进行加标回收的确定,其回收率必须达到方法和规范的要求,不能进行加标回收的可以用有证标准物质代替。
本实验对样品进行加标测试,加标量分别为0.25ug和0.75ug,重复测定6次,测定结果如下表:间,都满足标准方法的要求。
011 土壤 氰化物和总氰化物的测定 分光光度法 -作业指导书

XX公司作业指导书土壤氰化物和总氰化物的测定分光光度法修订页1编制依据本方法依据《土壤氰化物和总氰化物的测定分光光度法》(HJ 745-2015)编制。
2适用范围本标准规定了测定土壤中氰化物和总氰化物的分光光度法。
本标准适用于土壤中氰化物和总氰化物的测定。
当样品量为10 g,异烟酸-巴比妥酸分光光度法的检出限为0.01 mg/kg,测定下限为0.04mg/kg;异烟酸-吡唑啉酮分光光度法的检出限为0.04 mg/kg,测定下限为0.16 mg/kg。
3 术语和定义下列术语和定义适用于本标准。
3.1氰化物cyanide是指在pH=4介质中,硝酸锌存在下,加热蒸馏能形成氰化氢的氰化物,包括全部简单氰化物(多为碱金属和碱土金属的氰化物)和锌氰络合物,不包括铁氰化物、亚铁氰化物、铜氰络合物、镍氰络合物和钴氰络合物。
3.2总氰化物total cyanide是指在pH<2磷酸介质中,二价锡和二价铜存在下,加热蒸馏能形成氰化氢的氰化物,包括全部简单氰化物(多为碱金属和碱土金属的氰化物,铵的氰化物)和绝大部分络合氰化物。
4方法原理4.1 异烟酸-巴比妥酸分光光度法试样中的氰离子在弱酸性条件下与氯胺T反应生成氯化氰,然后与异烟酸反应,经水解后生成戊烯二醛,最后与巴比妥酸反应生成紫蓝色化合物,该物质在600 nm 波长处有最大吸收。
4.2 异烟酸-吡唑啉酮分光光度法试样中的氰离子在中性条件下与氯胺T反应生成氯化氰,然后与异烟酸反应,经水解后生成戊烯二醛,最后与吡唑啉酮反应生成蓝色染料,该物质在638 nm 波长处有最大吸收。
5 干扰和消除当试样微粒不能完全在水中均匀分散,而是积聚在试剂-空气表面或试剂-玻璃器壁界面时,将导致准确度和精密度降低,可在蒸馏前加5 ml 乙醇以消除影响。
试样中存在硫化物会干扰测定,蒸馏时加入的硫酸铜可以抑制硫化物的干扰。
试料中酚的含量低于500 mg/L 时不影响氰化物的测定。
油脂类的干扰可在显色前加入十二烷基硫酸钠予以消除。
土壤 氰化物和总氰化物的测定 分光光度法(HJ 745-2015)

4.1 异烟酸-巴比妥酸分光光度法
1
试样中的氰离子在弱酸性条件下与氯胺T反应生成氯化氰,然后与异烟酸反应,经水解后生 成戊烯二醛,最后与巴比妥酸反应生成紫蓝色化合物,该物质在600 nm 波长处有最大吸收。 4.2 异烟酸-吡唑啉酮分光光度法
试样中的氰离子在中性条件下与氯胺T反应生成氯化氰,然后与异烟酸反应,经水解后生成 戊烯二醛,最后与吡唑啉酮反应生成蓝色染料,该物质在638 nm 波长处有最大吸收。
5 干扰和消除
当试样微粒不能完全在水中均匀分散,而是积聚在试剂-空气表面或试剂-玻璃器壁界面 时,将导致准确度和精密度降低,可在蒸馏前加5 ml 乙醇以消除影响。
试样中存在硫化物会干扰测定,蒸馏时加入的硫酸铜可以抑制硫化物的干扰。 试料中酚的含量低于500 mg/L 时不影响氰化物的测定。 油脂类的干扰可在显色前加入十二烷基硫酸钠予以消除。
称取 1.5 g 异烟酸(C6H6NO2)溶于 25 ml 氢氧化钠溶液(6.14)中,加水稀释定容至100ml。 6.16.2 吡唑啉酮溶液。
称取 0.25 g 吡唑啉酮(3-甲基-1-苯基-5-吡唑啉酮,C10H10ON2)溶于 20 ml N,N-二甲基甲酰 胺[HCON(CH3)2]中。 6.16.3 异烟酸-吡唑啉酮溶液。
2
6.10 氢氧化钠溶液:ρ(NaOH)=15 g/L。 称取 15.0 g 氢氧化钠溶于水中,稀释至1000 ml,摇匀,贮于聚乙烯容器中。
6.11 氯胺 T 溶液:ρ(C7H7ClNNaO2S·3H2O)=10 g/L。 称取1.0 g 氯胺T溶于水中,稀释至100 ml,摇匀,贮存于棕色瓶中,临用时现配。
6 试剂和材料
除非另有说明,分析时均使用符合国家标准的分析纯试剂,实验用水为新制备的蒸馏水或去 离子水。 6.1 酒石酸溶液:ρ(C4H6O6)=150 g/L。
土壤中有害氰化物的检测方法_韩康芹张云肖冯敏英

土壤中有害氰化物的检测方法韩康芹,张云肖,冯敏英(河北水文工程地质勘察院,河北石家庄050021)摘要[目的]为了准确检测出土壤中氰化物的含量。
[方法]《生活饮用水标准检验方法》GB /T 5750-2006异烟酸-吡唑酮分光光度法检测水中氰化物的方法已被广泛采用,但是土壤基质相当复杂,测定过程中基质干扰、固体颗粒的吸附、土样保存、蒸馏、萃取等过程均有可能导致其含量的损失,直接影响检测结果的准确性。
[结果]质量控制中,采用平行样品检测控制其精密度,加标回收率的测定控制其准确度。
[结论]检出结果能正确地反映氰化物在土壤中的含量。
关键词土壤;氰化物;检测方法;标准溶液中图分类号S156文献标识码A 文章编号0517-6611(2014)03-00729-02Detection Method of Harmful Canide in Soil HAN Kang-qin et al (Hebei Hydrogeological and Engineering Geological Exploration Institute ,Shijiazhuang ,Hebei 050021)Abstract [Objective ]The research aimed to accurately detect the content of cyanide in soil.[Method ]Isonicotinic acid-pyrazolone detec-tion spectrophotometry in Drinking Water Standard Test Method GB /T 5750-2006had been widely used in the detection of cyanide in water.The soil matrix was quite complex.Matrix interference in the process of determination ,solid particle adsorption ,soil conservation ,distillation ,and extraction processes were likely to lead to the content of the loss ,directly affect the accuracy of test results.[Result ]The quality was con-trolled by the determination of samples with parallel detection ,and the accuracy was controlled by the determination of recovery rate.[Conclu-sion ]The detection result was proved to correctly reflect the content of cyanide in soil.Key words Soil ;Cyanide ;Detection method ;Standard solution作者简介韩康芹(1965-),女,河北石家庄人,高级工程师,从事水、土检测工作。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
土壤中總氰化物檢測方法NIEA S411.60B一、方法概要於酸性條件下,土壤中氰化物在迴流蒸餾過程中反應成氫化氰(Hydrogen cyanide)後釋出,以氫氧化鈉溶液吸收後,可用比色法或滴定法測定氰化物濃度。
在比色法中,吸收溶液於pH<8時,氰離子與氯胺-T(Chloramine-T)反應轉換成氯化氰(CNCl),續與異菸鹼酸(4-pyridine carboxylic acid)及1,3-二甲基巴比妥酸(1,3-dimethylbarbituric acid)反應產生有色錯合物,使用分光光度計在波長606 nm處測其吸光度;在滴定法中,以硝酸銀溶液滴定吸收溶液中之氰離子,形成可溶之Ag(CN)2- 錯離子,使用對銀離子敏感之二甲胺基苯叉羅丹寧(5-(4-dimethylamino benzylidene) rhodanine)指示劑,達滴定終點時,溶液由黃色轉為橙紅色。
二、適用範圍(一) 本方法適用於未經風乾研磨處理之土壤、底泥等類似基質中總氰化物之檢測。
(二) 比色法適用於田間含水土壤中總氰化物含量0.5~50 mg/kg之樣品檢測;滴定法適用於田間含水土壤中總氰化物含量大於50 mg/kg之樣品檢測,若吸收液呈現混濁或有顏色時,稀釋後視需要用比色法或滴定法檢測。
(三) 本實驗之樣品及廢液屬氰系急毒性物質,相關安全措施及應注意事項如註1。
三、干擾二價錫及銅的鹽類可以抑制硫化物的干擾及促進氰化物錯合物的分解。
四、設備及材料(一) 蒸餾設備(含抽氣裝置):如圖一或具相同功能之設備。
(二) 分光光度計:使用波長606 nm,附1 cm光徑之樣品槽。
(三) 分析天平:精秤至0.1 mg。
(四) 滴定管:最小刻度0.05 mL。
(五) 磁石、電磁攪拌器。
(六) 滴定用燒杯:250 mL以下。
(七) 定量瓶。
五、試劑所有檢測時使用的試劑化合物除非另有說明,否則必須是分析級試藥。
(一) 蒸餾試劑1、試劑水:比電阻≧16 MΩ-cm且不含氰化物之純水。
2、氫氧化鈉溶液,1 M:溶解40 g氫氧化鈉於試劑水中,並以試劑水稀釋至1 L,儲存於塑膠瓶中。
3、乙醇:95%。
4、硫酸銅溶液:溶解200 g硫酸銅(CuSO4·5H2O)於試劑水中,並以試劑水稀釋至1 L。
5、鹽酸溶液,1M:取8.3 mL濃鹽酸於試劑水中,並以試劑水稀釋至100 mL。
6、氯化亞錫溶液:溶解5 g氯化亞錫(SnCl2·2H2O)於40 mL 1 M鹽酸溶液,並以試劑水稀釋至100 mL,本溶液須每日配製。
7、濃磷酸:85%。
(二) 比色法試劑1、氫氧化鈉溶液,0.8 M:溶解32 g氫氧化鈉於試劑水中,並以試劑水稀釋至1 L,儲存於塑膠瓶中。
2、對硝基酚(p-nitrophenol)溶液,0.1%:溶解0.1 g對硝基酚於100 mL乙醇。
3、醋酸溶液,20%(v/v):取100 mL冰醋酸(Glacial acetic acid),以試劑水稀釋至500 mL。
4、氯胺-T溶液:溶解0.5 g氯胺T(Chloramine-T trihydrate)於試劑水,並以試劑水稀釋至50 mL,本溶液須每日配製。
5、比色法呈色試劑:溶解7 g氫氧化鈉於約500 mL試劑水,加入16.8 g 1,3-二甲基巴比妥酸及13.6 g異菸鹼酸,以試劑水稀釋至1000 mL。
在30℃下混合1小時,保存於暗處10℃以下,使用前以8μm濾紙(Whatman 40或同級品)過濾。
(三) 滴定法試劑1、羅丹寧指示劑:溶解0.02 g二甲胺基苯叉羅丹寧於丙酮中,並以丙酮稀釋至100 mL,本溶液貯存於暗處室溫條件下,可保存一星期。
2、氯化鈉標準溶液,0.02 M:溶解1.1688 g之一級標準品級氯化鈉(先於105℃乾燥隔夜)於試劑水,並以試劑水稀釋至1000 mL。
3、鉻酸鉀指示劑:溶解5.0 g鉻酸鉀(K2CrO4)於少量試劑水,加入硝酸銀溶液直至生成紅色之沉澱;靜置12小時,過濾,然後以試劑水稀釋至100 mL。
4、硝酸銀溶液,0.01 M:溶解1.6897 g硝酸銀於約400 mL試劑水,並以試劑水稀釋至1000 mL,貯存於棕色玻璃瓶並置於暗處,本溶液每兩星期用氯化鈉標準溶液標定,標定方法如下:標定硝酸銀溶液:取氯化鈉標準溶液10.0 mL,以試劑水稀釋至100 mL,以1N氫氧化鈉溶液調整其pH至7至8,加入1.0 mL鉻酸鉀指示劑以硝酸銀溶液滴定至帶桃紅色之黃色終點,同時以試劑水作空白試驗,依下式計算硝酸銀溶液濃度:M:氯化鈉標準溶液濃度(M)。
A:氯化鈉標準溶液消耗硝酸銀溶液之體積(mL)。
B:空白試驗消耗硝酸銀溶液之體積(mL)。
(四) 氰化物儲備溶液,100 mg CN-/L:溶解250 mg氰化鉀於0.8 M氫氧化鈉溶液中,並以0.8 M氫氧化鈉溶液稀釋至1000 mL,儲存於10℃以下,每日以0.01 M之硝酸銀溶液標定,標定方法如下,或者亦可使用市售經確認之標準溶液。
精取20.0 mL氰化物儲備溶液,加入1.0 mL羅丹寧指示劑溶液後,以已知莫耳濃度之硝酸銀溶液滴定至顏色由黃色變橙紅色,即為滴定終點;同時以0.8 M氫氧化鈉溶液執行空白試驗。
依下式計算氰化物儲備溶液之氰離子濃度:C:氰化物儲備溶液消耗硝酸銀溶液之體積(mL)。
D:空白試驗消耗硝酸銀溶液之體積(mL)。
M:硝酸銀溶液濃度(M)。
V:氰化物儲備溶液體積(mL)。
(五) 氰化物標準溶液,10 mg CN-/L:取10.0 mL氰化物儲備溶液,以0.8 M氫氧化鈉溶液稀釋至100 mL,本溶液須每日配製。
六、採樣與保存土壤、底泥等類似基質樣品之採樣依據「土壤採樣方法(NIEA S102)」採集具有代表性之土壤樣品約500 g,如含氣態氰化物,則依揮發性有機物樣品採樣方式採樣。
儲存於塑膠或玻璃容器中,樣品冷藏於4 ±2℃暗處,並於採樣後14天內進行分析。
七、步驟(一) 樣品前處理1、取樣檢測前勿打開樣品瓶,如為均質樣品,取樣時應儘速取出所需樣品量;如果樣品無法混合均勻,則須執行重複樣品分析。
2、秤取適量田間含水土壤樣品(註2),依照「土壤水分含量測定方法-重量法」(NIEA S280)測定土壤中水分含量。
3、同時另取10.0 g土壤樣品置於蒸餾瓶中,加入約160 mL試劑水(註3)確認進氣管沒入蒸鎦瓶液面下。
4、取40 mL 1 M氫氧化鈉溶液置於氣體吸收管,連接蒸餾裝置如圖一。
5、打開抽氣泵,由進氣管加入10 mL硫酸銅溶液及2 mL氯化亞錫溶液,以抑制硫化物的干擾及催化氰化物錯合物之分解,以試劑水沖洗管壁,保留少量試劑水在進氣管中以維持氣密。
調整微調控制閥以控制抽氣泵至適當抽氣速率(例如15 L/小時),確保所釋出之氰化氫被帶入氣體吸收管中。
6、由進氣管加入20 mL濃磷酸,以6 mL以上的試劑水沖洗管壁,保留2~3 mL試劑水在進氣管中以維持氣密。
7、打開加熱裝置並和緩迴流120 ±10分鐘。
8、停止加熱並繼續抽氣15分鐘後,緩慢打開進氣管之栓塞以釋除蒸餾瓶內半真空(Partial vacuum),注意若未緩慢釋除蒸餾瓶內半真空,將導致氣體吸收瓶內之氫氧化鈉回抽至蒸餾瓶中。
9、將氫氧化鈉吸收液倒入50 mL定量瓶,以5 mL以上的試劑水淋洗連接管及氣體吸收管,洗液併入上述量瓶中,以試劑水稀釋至刻度後,保留於10℃暗處待分析。
(二) 比色法1、樣品檢測(1) 取20.0 mL吸收液置於50 mL定量瓶,加入2滴對硝基酚溶液,混合均勻後,邊混合邊逐滴加入20%醋酸溶液直至溶液由黃色變成無色。
(2) 加入2 mL氯胺-T溶液,蓋上瓶塞靜置5 ±1分鐘(淡黃色)。
(3) 加入6 mL呈色試劑,混合均勻後(淡黃色變藍色)以試劑水稀釋至刻度。
(4) 於加入呈色試劑後20 ±5分鐘,以分光光度計在波長606 nm處測定吸光度。
(5) 若吸收液中氰化物濃度大於1.0 mg/L,應減少吸收液取用量,再重複(1)-(4)。
2、檢量線製備取氰化物標準溶液10 mg/L,配製一個空白和至少五種不同濃度的檢量線標準溶液,例如取0.5、1.0、2.0、4.0、5.0 mL,以0.8 M氫氧化鈉溶液使體積約為20 mL,依步驟七、(二)2操作並讀取吸光度,以標準溶液濃度(mg/L)為X軸,吸光度為Y軸,繪製一吸光度與氰化物濃度﹙mg/L﹚之檢量線,其濃度為0、0.1、0.2、0.4、0.8、1.0 mg / L。
(三) 滴定法1、取20 mL吸收液置於50 mL燒杯,加入0.5 mL滴定法指示劑,以硝酸銀溶液滴定至顏色由黃色變橙紅色(註4)。
2、如果以0.01 M硝酸銀溶液滴定的體積超過10 mL,取另一份吸收液,改以0. 1 M硝酸銀溶液滴定。
若此時硝酸銀溶液滴定的體積仍超過10 mL,則減少吸收液取樣體積並重新滴定。
八、結果處理(一) 依下式計算由比色法得出之氰化物濃度A:檢量線求得之氰化物濃度(mg/L)。
V:樣品前處理後,上機檢測樣品之體積50 mL。
f:上機測試時之稀釋倍數。
(如吸收液取20 mL比色時f=2.5)W:土壤取樣重(g)。
(土壤水分含量以乾基為準)。
基(二) 依下式計算由滴定法得出之氰化物濃度A:滴定時吸收液消耗硝酸銀溶液之體積(mL)。
B:空白試驗消耗硝酸銀溶液之體積(mL)。
M:硝酸銀溶液濃度(M)。
V1:樣品前處理後吸收液之最終定量體積50 mL。
V2:滴定時取用之吸收液體積(mL)。
W:土壤取樣重(g)。
(土壤水分含量以乾基為基準)。
九、品質管制(一) 比色法檢量線:每次樣品分析前應重新製作檢量線,其線性相關係數(r值)應大於或等於0.995。
檢量線製作完成應即以第二來源標準品配製接近檢量線中點濃度之標準品確認,相對誤差值應在±15%以內。
(二) 檢量線查核:每批次或每10個同一來源樣品分析結束時,應再執行一次檢量線查核,以檢量線中間濃度附近的標準溶液進行查核,相對誤差值應在±15%以內。
(三) 空白樣品分析:每批次或每20個同一來源樣品應至少再執行一次空白樣品分析,空白樣品分析值應小於二倍方法偵測極限。
(四) 重複樣品分析:每批次或每20個同一來源樣品應至少再執行一個重複樣品分析,其相對差異百分比應在25%以內。
(五) 添加樣品分析:每20個同一來源樣品應執行一個添加樣品分析,若每批次樣品數少於20個,則每批次仍應執行一個添加樣品分析,其添加回收率應在75~125%。
(六) 查核樣品分析:每40個同一來源樣品應併同分析一個參考標準樣品(至少為CRM等級),若每批次樣品數少於40個,則每批次仍應執行一個參考標準樣品分析,其回收率應在70~130%。