利用电位滴定法进行含量测定
含量测定分析方法

含量测定分析方法含量测定分析方法是化学分析中常用的一种分析方法,用于确定样品中某种化学物质的含量或浓度。
根据不同的化学物质和样品性质,含量测定分析方法可以有多种不同的选择,下面将介绍几种常见的含量测定分析方法。
一、滴定法滴定法是一种将标准溶液溶液逐滴加入待测物溶液中,通过标准溶液与待测物发生化学反应达到等价点来确定待测物含量的方法。
滴定法适用于有明确反应产物生成的物质,例如酸碱滴定法、络合滴定法等。
滴定法通常需要使用酸碱指示剂来标示化学反应的等价点,指示剂的颜色变化可以帮助确定滴定终点。
二、分光光度法分光光度法是通过测量样品溶液在特定波长光线下的透过率或吸光度来确定样品中某种物质的含量。
分光光度法适用于有明显吸收峰的物质,例如红外吸收光谱、紫外可见吸收光谱等。
分光光度法通常需要建立标准曲线,根据光强与物质浓度之间的线性关系来计算待测物的含量。
三、电位滴定法电位滴定法是利用电位计测定待测物溶液的电位变化来确定物质含量的方法。
电位滴定法适用于有明确电位变化的化学反应,例如氧化还原滴定法。
在氧化还原滴定中,待测物与滴定剂发生氧化还原反应,通过监测电位的变化来确定滴定终点。
四、火焰原子吸收光谱法火焰原子吸收光谱法是利用待测物在火焰中产生的原子吸收特性来确定元素含量的方法。
火焰原子吸收光谱法适用于分析金属元素的含量,例如钠、铜、铁等。
通过将样品溶解在溶剂中,喷入预热的火焰中,测量样品溶液对特定波长的光的吸收程度,从而计算待测元素的含量。
五、高效液相色谱法高效液相色谱法是一种基于分配与吸附原理的分析方法,通过样品在固定填料和流动相作用下的相互分离来确定物质的含量。
高效液相色谱法适用于分析有机物的含量,例如药物、环境污染物等。
通过选择合适的固定相、流动相以及检测器,将待测物与其他组分分离,并根据谱图来计算待测物的含量。
以上所述只是常见的几种含量测定分析方法,实际上还有很多其他的测定方法,如电感耦合等离子体发射光谱法、原子荧光光谱法、电化学法等。
实验5 电位滴定法测定醋酸的含量

实验5 电位滴定法测定醋酸的含量一、实验目的1.了解电位滴定的原理和方法;3.熟练制备标准物质溶液;4.掌握化学计量学原理及方法。
二、实验原理电位滴定法是一种利用电位滴定仪对被测物质进行定量分析的方法。
电位滴定仪是用电极法律测定被测溶液中的化合物数量浓度的轻便实验仪器。
它利用滴定试液溶质参与电位反应而容易确定终点。
电位滴定法因效果准确、反应灵敏快捷、操作简单等特点,广泛应用于分析化学中。
2.醋酸含量测定原理醋酸是一种有机酸。
在一定浓度的醋酸酸性溶液中,用强碱性草酸钠溶液作标准滴定溶液,可以用电位滴定法测定醋酸的酸度。
如下所示:CH3COOH+Na2C2O4 (标准滴定溶液)=CH3COO-Na+(CH2(COOH)(COONa))用紫酸钾作为指示剂,草酸钠溶液的滴定终点为pH8.1,滴定过程中,试剂反应快,终点尖锐,误差小。
三、实验步骤1.制备0.1mol/L的草酸钠溶液称取4.15g草酸钠(粉末)于1000mL容量瓶中,加入水溶液至刻度,混匀即为0.1mol/L 的草酸钠溶液。
草酸钠不易溶于水,搅拌约30min即可。
实验时用0.1mol/L草酸钠溶液作为标准滴定溶液浸泡电极。
称取6.0mL稀醋酸于100mL容量瓶中,加入水至90mL,混匀后加入0.1mol/L的HCl溶液至刻度,摇匀即为0.1mol/L醋酸溶液。
千万不要把HCl加入稀醋酸中,因为稀醋酸很容易挥发,加了HCl后,稀醋酸会剧烈挥发,造成浓度偏低。
3.进行电位滴定(1)置入电极法化电极的三个滴头分别放入试剂瓶、标准溶液瓶和样品瓶中,将电极泡“(+)-AgCl”能接触溶液的电极头首先摆放于标准溶液瓶中。
(2)标定电极用0.1mol/L的草酸钠溶液标定电极,使其显示读数为0。
(3)电极反应将电极头和试剂瓶中的紫色铁钾指示剂之间挤入一滴草酸钠溶液,熟练搅拌、静置,使电极反应平衡。
读出电极的电动势值。
(4)样品反应将电极头首先摆放于样品瓶中,将0.1mol/L醋酸溶液滴入到试管中,50-100mL左右,滴入草酸钠溶液后,灵敏搅拌,使其反应均匀稳定,使草酸钠溶液与醋酸中的氢离子反应完全,每滴要搅匀,直到铁钾指示剂由紫变褐即结束反应。
电位滴定法与手动滴定法测定药品含量的比较

电位滴定法与手动滴定法测定药品含量的比较作者:梁永革李丽来源:《中国现代医生》2017年第19期[摘要] 目的对比电位滴定法与手动滴定法在测定药品含量方面的效果。
方法将我区医疗机构常用药物阿司匹林、盐酸左旋咪唑、氯化钠注射液作为研究对象,分别应用电位滴定法与手动滴定法对上述药品含量进行测定,并对不同检测方法下阿司匹林、盐酸左旋咪唑、氯化钠注射液三类药品的定量检测结果及相对标准偏差进行对比分析。
结果对比两种检测方法下的定量检测结果,电位滴定法下对阿司匹林、盐酸左旋咪唑、氯化钠注射液的检测结果与手动电位滴定法无明显差异(P>0.05)。
两种检测方法下的相对标准偏差对比,电位滴定法下对阿司匹林、盐酸左旋咪唑、氯化钠注射液的相对标准偏差均显著低于手动滴定法,差异有统计学意义(P[关键词] 药品含量;电位滴定;手动滴定;定量检测[中图分类号] R927.2 [文献标识码] B [文章编号] 1673-9701(2017)19-0040-03[Abstract] Objective To compare the effect of potentiometric titration and manual titration on the determination of drug content. Methods Aspirin, levamisole hydrochloride and sodium chloride injection which were commonly used in our district were selected as the subjects. Potentiometric titration and manual titration were applied respectively for the determination of the contents of the above drugs. The quantitative analysis results and the relative standard deviations of the three types of drugs of aspirin, levamisole hydrochloride and sodium chloride injection were compared and analyzed under different determination methods. Results The quantitative determination results were compared under the two determination methods, and there was no significant difference between the determination results of aspirin, levamisole hydrochloride and sodium chloride injection under the potentiometric titration and manual titration methods, and the difference was not statistically significant (P>0.05). The relative standard deviation was compared under the two determination methods, and the relative standard deviation of aspirin, levamisole hydrochloride and sodium chloride injection under the potentiometric titration was significantly lower than that under manual titration, and the difference was statistically significant(P[Key words] Drug contents; Potentiometric titration; Manual titration; Quantitative determination滴定分析是目前化學定量分析领域中应用最为广泛的技术手段之一[1-3]。
自动电位滴定仪测定银含量

七,总结
三个批次纳米银溶液中的银含量范围在0.12±0.01%。Mettler Toledo自动 滴定仪,在滴定过程中可以自动绘出滴定曲线,自动找出滴定终点,自动给出 体积及计算结果,果,重复性及准确率高,可以满足银含量的检测需求。
六,实验数据
1.纳米银溶液批次S20210708002
①VNaCl=1.017mL,mSnw=5.1945g,
②VNaCl=1.220mL,mSnw=5.8220g, ③VNaCl=1.297mL,mSnw=6.1011g,
C' Ag 0.05003 1.017 107.8682 100 0.1056% 5.1945 1000
C' Ag 0.05003 1.220 107.8682 100 0.1131% 5.8220 1000
C' Ag 0.05003 1.297 107.8682 100 0.1147% 6.1011 1000
2.纳米银溶液批次S20210708004
①VNaCl=1.369mL,mSnw=6.3412g,
C' Ag 0.05003 1.304 107.8682 100 0.1209% 5.8180 1000
C' Ag 0.05003 1.294 107.8682 100 0.1271% 5.4915 1000
C' Ag 0.05003 1.304 107.8682 100 0.1240% 5.6729 1000
四,计算公式
银离子计算公式为:
C' Ag cNacl VNacl MAg 100 mSnw 1000
C'Ag+——样品中银离子的含量,%; cNacl——基准试剂氯化钠溶液浓度,单位为 (mol/L); VNaCl——待测液中银离子所消耗氯化钠溶液 体积,单位为(mL); MAg——银的摩尔质量,单位为(g/mol);
实验5 电位滴定法测定醋酸的含量
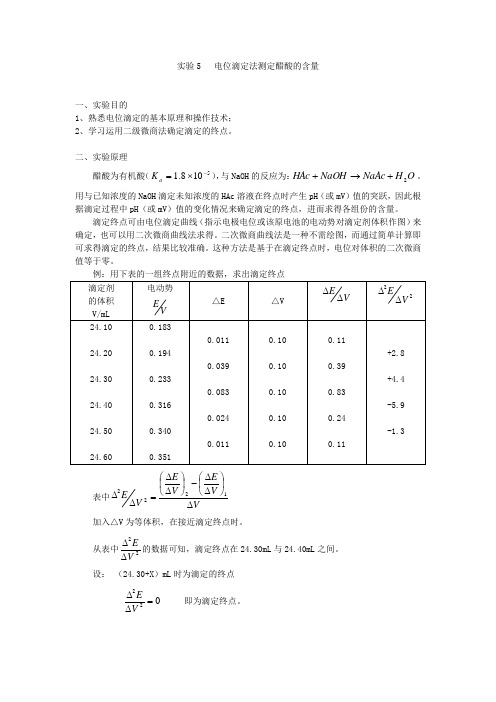
实验5 电位滴定法测定醋酸的含量一、实验目的1、熟悉电位滴定的基本原理和操作技术;2、学习运用二级微商法确定滴定的终点。
二、实验原理醋酸为有机酸(5108.1-⨯=a K ),与NaOH 的反应为:O H NaAc NaOH HAc 2+→+。
用与已知浓度的NaOH 滴定未知浓度的HAc 溶液在终点时产生pH (或mV )值的突跃,因此根据滴定过程中pH (或mV )值的变化情况来确定滴定的终点,进而求得各组份的含量。
滴定终点可由电位滴定曲线(指示电极电位或该原电池的电动势对滴定剂体积作图)来确定,也可以用二次微商曲线法求得。
二次微商曲线法是一种不需绘图,而通过简单计算即可求得滴定的终点,结果比较准确。
这种方法是基于在滴定终点时,电位对体积的二次微商值等于零。
例:用下表的一组终点附近的数据,求出滴定终点 滴定剂 的体积 V/mL 电动势VE△E △V VE∆∆22V E∆∆24.10 24.20 24.30 24.40 24.50 24.600.183 0.194 0.233 0.316 0.340 0.3510.011 0.039 0.083 0.024 0.0110.10 0.10 0.10 0.10 0.100.11 0.39 0.83 0.24 0.11+2.8 +4.4 -5.9 -1.3表中VV E V E V E ∆⎪⎭⎫ ⎝⎛∆∆-⎪⎭⎫ ⎝⎛∆∆=∆∆1222 加入△V 为等体积,在接近滴定终点时。
从表中22VE∆∆的数据可知,滴定终点在24.30mL 与24.40mL 之间。
设: (24.30+X )mL 时为滴定的终点022=∆∆VE即为滴定终点。
则有:02212=∆∆=∆⎪⎭⎫ ⎝⎛∆∆-⎪⎭⎫ ⎝⎛∆∆V EV V E V E +++-+⎪⎪⎭⎫ ⎝⎛∆∆⨯⎪⎪⎭⎫ ⎝⎛∆∆-⎪⎪⎭⎫ ⎝⎛∆∆-+=22-2222V E V E V E V V V V 终 即: ()[]4.49.54.430.2440.24+24.3=V 终⨯---所以在滴定终点时滴定剂的体积应为: )(34.24mL V =终三、仪器和试剂1、仪器 酸度计(含复合电极) 电磁搅拌器(含搅拌子) 滴定管100µL 进样器铁架台(含滴定管夹) 2、试剂 邻苯二甲酸氢钾NaOH 溶液0.1mol/L :称取4g 固体NaOH ,加入新鲜的或煮沸的除去二氧化碳的蒸馏水,完全溶解后,定容至1L ,充分摇匀(待标定)。
实验45 电位滴定法连续测定工业废水中碘和氯的含量

2. 滴定
五、实验数据及处理
电位滴定终点确定方法
(1) E-V 曲线法
如图( a)所示: E-V 曲线法简单,但准确 性稍差。
(2) ΔE/ΔV - V 曲线法
如图(b)所示。 由电位改变量与滴定剂体积 增量之比计算之。 ΔE/ΔV - V曲线上存在着极值点,该点对应着
七、思考题
在滴定试液中加入Ba(NO3)2的目的是什么? 因为卤化银沉淀易吸附溶液中的银离子和卤素 离子而带来误差,在试液中加入硝酸钡,由于碘化 银沉淀能吸附浓度较大的硝酸根或钡离子,因而减 少了对银离子的吸附作用而使误差减小。
如图( a)所示: E-V 曲线法简单,但准确 性稍差。
(2) ΔE/ΔV - V 曲线法
如图(b)所示。 由电位改变量与滴定剂体积 增量之比计算之。 ΔE/ΔV - V曲线上存在着极值点,该点对应着
E-V 曲线中的拐点。
(3) Δ2E/ΔV
2
- V 曲线法
Δ2E/ΔV 2表示E-V 曲线的二阶微商。 Δ2E/ΔV 2值由下式计算:
E E )2 ( )1 E V V 2 V V
2
(
滴定反应为: Ag++Cl-→AgCl↓ Ksp=1.8×10-10 Ag++I-→AgI↓ Ksp=8.3×10-17 化学计量点时,[Ag+]=[Cl-]或[Ag+]=[I-], 可由KspAgCl或KspAgI求出Ag+的浓度,由此计算出Ag 电极的电位。
E-V 曲线中的拐点。
(3) Δ2E/ΔV
2
- V 曲线法
Δ2E/ΔV 2表示E-V 曲线的二阶微商。 Δ2E/ΔV 2值由下式计算:
自动电位滴定法测定fe2+实验报告

自动电位滴定法测定fe2+实验报告
实验目的:使用自动电位滴定法测定Fe2+的浓度。
实验原理:自动电位滴定法是一种电化学分析方法。
它利用电流与电势之间的关系,通过向溶液中滴加标准溶液,控制电位变化来测定待测溶液中的物质含量。
实验步骤:
1. 预处理:将所需玻璃仪器清洗干净,使其干燥;
2. 准备标准溶液:称取0.025mol/L的KMnO4溶液和0.2mol/L的
H2SO4溶液,混合,用去离子水定容至1L,摇匀;
3. 将待测Fe2+溶液和标准KMnO4溶液各取10mL,加入预处理好的电极中,同时加入1~2mL的三乙胺,开始滴定;
4. 在滴定过程中,记录滴定电压和溶液体积,直至滴定结束;
5. 用公式计算Fe2+的浓度:C(Fe2+)= (V2 –V1)×C1/V1×n,其中V1为待测溶液体积,C1为KMnO4溶液浓度,V2为滴定终点体积,n 为KMnO4溶液中含有的Fe2+的当量数。
实验结果:对10 mL的待测Fe2+溶液滴加了9.8 mL的
0.025mol/L KMnO4溶液,计算得到Fe2+的浓度为:C(Fe2+)= (9.8-0)×0.025/0.01×5 = 12.25mol/L。
实验结论:本实验使用自动电位滴定法测定了Fe2+的浓度,可得到较准确的结果。
但需注意滴定过程中要控制好滴液速度,以免出现误差。
自动电位滴定测定混合碱的组分及含量

自动电位滴定测定混合碱的组分及含量自动电位滴定法是一种通过测量滴定过程中电极电位变化来确定被测物质浓度的方法。
对于混合碱的测定,通常采用酸碱滴定法,利用酸与碱的中和反应来确定各自的含量。
而自动电位滴定法可以更准确地测定混合碱的组分及含量,以下是测定步骤:一、实验原理在滴定过程中,随着酸或碱的加入,溶液的pH值发生变化。
而电极电位与溶液的pH值存在一定的关系,因此可以通过测量电极电位的变化来监测滴定过程。
当加入的酸或碱恰好与混合碱中的一种成分完全反应时,溶液的pH值会发生突变,此时电极电位也会发生明显的变化。
通过自动滴定系统,可以准确地控制酸的加入量,并记录电极电位的变化,从而确定混合碱中各组分的含量。
二、实验步骤1.准备好实验所需试剂和仪器,包括混合碱样品、酸碱指示剂、电极、滴定管、磁力搅拌器等。
2.将电极放入滴定管中,加入适量的去离子水,开启磁力搅拌器,记录此时的电极电位。
3.用移液管准确移取一定体积的混合碱溶液,加入到滴定管中,记录此时的电极电位。
4.开启滴定管,加入适量的已知浓度的盐酸,使混合碱溶液中的一种组分完全反应。
在滴定过程中,电极电位会发生明显的变化。
5.继续加入适量的已知浓度的氢氧化钠溶液,使另一种组分完全反应。
在滴定过程中,电极电位会再次发生明显的变化。
6.根据电极电位的变化和加入的酸的体积,计算出第一种组分的含量;同理,根据第二次滴定的电极电位变化和加入的氢氧化钠的体积,计算出第二种组分的含量。
7.重复以上步骤,测试不同的混合碱样品,以了解各组分的含量变化情况。
三、实验结果分析通过自动电位滴定法测定混合碱的组分及含量,具有较高的准确性和可靠性。
在实验过程中,需要注意以下几点:1.实验前需对所用试剂的浓度、质量等进行仔细检查,以保证实验结果的准确性。
2.在滴定过程中,要保证滴定管干净,避免残留物对实验结果的影响。
3.在计算含量时,需要结合已知化学反应方程式和指示剂变色点进行计算,同时也要考虑溶液的密度等因素。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
利用电位滴定法进行含量滴定
1.相关定义及其用途
⑴电位滴定法主要用于容量分析确定终点或帮助确定终点。
⑵ 对于观察终点很不方便的外指示剂法和某些必须过量滴定液才能使指
示剂到达终点的容量分析法,采用电位或永停滴定法能使结果更加准确。
⑶ 我们把在非水溶剂中进行滴定的分析方法叫做非水滴定法。
非水溶剂指的
是有机溶剂与不含水的无机溶剂。
以非水溶剂作为滴定介质,不仅能增大有机化合物的溶解度,而且能改变物质的化学性质,使在水中不能进行完全的滴定能够顺利进行,从而扩大了滴定分析的应用范围。
2.仪器和性能要求
⑴电位滴定仪及自动电位滴定仪主要用于确定滴定终点,带有电位测定部
分的 PH计也可满足要求。
⑵电极玻璃电极为指示电极,饱和甘汞电极为参比电极
3.试药与试液
⑴滴定液
配置、标定与贮藏均应按药典规定;酸碱滴定液的标定,应不得少于三
分,酸滴定液标定和复标的RSD≤0.1%, 碱滴定液标定和复标的RSD≤
0.2%
⑵试液及试剂
醋酸汞试液及非水溶液滴定用的各种指示液均按药典规定配置;非水溶
液滴定用试剂的含水量应为0.01~ 0.02%
4.含量测定操作方法
⑴第一法用高氯酸液(0.1mol/l)滴定碱液药物
对高氯酸进行标定→空白效正→精密称取供试品(约消耗滴定液8ml)
→10 ~ 30ml 冰醋酸溶解→ 1 ~ 2 滴指示剂→高氯酸滴定并记录滴定过程中
消耗的滴定液体积 V、电位 E,以及颜色变化(注:当电位变化较大时,应
减慢滴定的速度,一般要滴至过量即至颜色不变为止)→ 处理数
据,(可通过 E-V 曲线法、E/ V - V曲线法或 2 E/V2 - V曲线
法)确定终点→ 计算供试品含量
⑵第二法用碱滴定液滴定酸性药物(方法与第一法相似)
5.记录与计算
⑴按规定记录称样量,滴定液标定时的温度、浓度,测定样品时的温度,
以计算滴定液的浓度;滴定液重新标定记录全部数据;记录滴定管的编
号、样品及空白试验,消耗的定液的读数及校正值。
⑵如滴定液需要重新标定,则应有标定滴定液的全记录。
⑶记录所用滴定管的编号、样品及空白试验,消耗滴定液的读数及校正值。
⑷计算:样品含量 % = (V1-V 2)× F× E× 100%
M
V 1 供试品消耗低定液的体积(ml)
V 2 空白试验消耗滴定液的体积(ml)
F为滴定液浓度的校正因子
E为滴定度( mg/ml)
M为称样量( mg)
6.滴定结果的判断
⑴供试品每次测定应在 2 份以上。
⑵原料药的高氯酸液直接滴定者,相对偏差不超过 0.15%,用碱滴定液直接
滴定者,相对偏差不超过 0.3%。
⑶若计算出的滴定终点与滴定的电位突变点一致,熟练后也可将电位突变
点时的指示剂颜色,作为以后含量滴定终点的颜色判断。
7.注意事项
⑴ 安全高氯酸滴定液可按药典规定的方法进行配置,其具有腐蚀性配置
时应注意防护。
冰醋酸有刺激性,高氯酸与有机物接触,遇热极易引起
爆炸,和醋酐混合时易发生剧烈反应放出大量热,因此配制高氯酸滴定液时,
应先将高氯酸用冰醋酸稀释后再在不断搅拌下缓缓滴加适量醋酐,量取高氯酸的量筒不得量醋酐,以免引起爆炸。
⑵ 水分由于水分的存在,将严重影响电位滴定曲线的突跃的指示剂颜
色的变化,影响终点的灵敏度,所有仪器,供试品中均不得有水分存在,所用的试剂的含水量均应在 0.2%以下,必要时应加入适量的醋酐以脱水。
冰醋酸在使用前,宜作空白试验,方法:取冰醋酸5~10ml 于50ml 锥形瓶中,加结晶紫指示液 1 滴,应为紫色,加高氯酸滴定液( 0.1mol/L )1 滴'' 即
应变为黄绿色,若为蓝色,则表示有水分存在,可加醋酐脱水,或加醋酐后重
蒸一次。
⑶指示剂不宜多加,以 1 ~ 2 滴为宜。
⑷温度变化对滴定介质冰醋酸影响较大,冰醋酸的凝点为15.6 ℃,当室
温低于 15.6 ℃,滴定液就会凝结在滴定管中,因此滴定温度应控制在 20℃
以上。
冰醋酸的膨胀系数较大,为 0.0011 ℃,即温度改变 1℃,体积就有 0.11% 的变化,所以当使用与标定温度相差在± 10℃以内,可根据下式将滴定液浓度加以校正 C1 =C0 / [ 1+0.0011(t 1- t 0)] ;如使用与标定温度相差在 10℃
以上,或滴定液放置一个月以上,使用时应重新标定。
如条件允许,可单独
安排或隔出一个房间,安装空调,作为非水溶液滴定室,标定溶液与测定供
试品在相同条件下进行,可避免温度影响,使测定结果更加准确。
⑸ 电极的选择电位滴定法:中和滴定常用玻璃电极;沉淀滴定常用银
电极;氧化还原滴定常用铂电极;非水溶液滴定采用玻璃电极和饱和甘汞电
极;永停滴定法:电极采用铂 - 铂电极。
(玻璃电极使用之前在冰醋酸中浸泡过夜;甘汞电极与玻璃电极使用前后均应用适当的溶剂清洗)
⑹滴定样品与标定高氯酸滴定液的温度差别超过10℃时,应重新标定
⑺ 控制温度、搅拌速度,以免影响测定结果。
⑻ 准确记录数据,为滴定终点的判断提供方便
⑼ 空白试验在所有的滴定中,均需同时另作空白试验,以消除试剂引
入的误差,尤其是在加醋酸汞试液的情况下。
⑽终点判定由于非水滴定法滴定终点的颜色变化复杂,对不同颜色的描述和感受也因人而异,因此终点判定以电位法为准,同时采用指示液以对照观察终点颜色的变化,待熟练掌握其颜色变化后,即可不必每次用电位法测定。
⑾贮存高氯酸滴定液应贮于棕色瓶中避光保存,若颜色变黄,即说明高氯酸部分分解,不得应用。