《遗传学》第十一章 分子病与先天性代谢缺陷
分子病与先天性代谢缺陷病

C.密码子插入或缺失:珠蛋白基因序列中插入或缺失1个或数个密 码子,发生读码顺序移动或使珠蛋白肽链增加或减少相应的1个或数个 氨基酸。
D.融合基因:同源染色体的不等交换,导致这些异常的血红蛋白链 由两种不同珠蛋白部分肽链连接而成。
控制珠蛋白肽链的α基因簇在16p13.2,人类二 倍体细胞中共有4个α基因;类β基因簇位于11p1 5.4。
(一) 血红蛋白病
2.异常血红蛋白病 (1)异常血红蛋白病的珠蛋白基因突变类型: A.单个碱基的取代:①错义突变 如(镰型细胞贫血症),系血红蛋
白β链第6位密码子由6(谷氨酸)突变为6(缬氨酸)。②无义突变 如 , 系血红蛋白β链第145位密码子由(酪氨酸)突变为终止密码子。③终止 密码子突变 如 ,由血红蛋白α链的终止密码子突变为(谷氨酰胺), 使α链非正常延长至172个氨基酸才终止。
(一) 血红蛋白病
(2)β 地中海贫血:①重型β 地中海贫血:患者正常β 链缺乏或合 成量很少,血红细胞中无或含量很少,和2含量增高,α 珠蛋白链过剩 而沉积到红细胞膜上,改变了红细胞膜的性能,引发严重的溶血性贫 血。主要临床症状表现为患儿出生几月后溶血反应,并伴有腹泻、发 热、生长发育迟缓、身材矮小、骨髓增生,可出现鼻塌眼肿、上颌前 突、头大额隆等特殊的“地中海贫血面容”。②中间型β 地中海贫血: 临床表现介于轻型和重型之间,中度贫血,脾脏轻或中度肿大,黄疸 可有可无,骨骼改变较轻。③轻型β 地中海贫血:带有一个正常的β 珠蛋白基因,一般无临床症状或有轻微贫血和脾脏肿大。
(四) 膜转运载体蛋白病
肝豆状核变性的遗传又称病,由编码P型铜转运酶的基因(7)突变引起, 基因定位于13q14.3,为常染色体隐性遗传,7基因突变多为错义突变 或无义突变。临床表现以神经系统和肝脏受损为主,晚期表现为痴呆。 患者可通过服期 治疗,因此本病的早期诊断对于临床治疗很有意义。
复旦大学上海医学院医学遗传学名词解释

医学遗传学名词解释第一章绪论1.medical genetics(医学遗传学)是用人类遗传学的理论和方法研究遗传病从亲代到子代的特点和规律、起源和发生、病理机制、病变过程及其与临床关系(诊断、治疗和预防)的一门综合性学科。
2.genetic disease(遗传病)细胞内的遗传物质在数量、结构和功能方面发生改变所引起的疾病。
其发生需要有一定的遗传基础;通过这种基础,能按一定方式传给后代。
在现代医学中,遗传病的概念有所扩大,逐渐强调环境因素所起的作用。
3.somatic cell genetic disorder(体细胞遗传病)是指只能在特异的体细胞中发生的遗传病,不能在世代间垂直传递。
体细胞基因突变是此类疾病发生的基础。
主要包括恶性肿瘤、白血病、自身免疫缺陷病、衰老等。
在经典的遗传病的概念中,并不包括此类疾病。
4.recurrence risk(再发风险率)是指病人所患的遗传病在家系亲属中再次发生的风险率。
第二章人类基因1.gene(基因)是DNA(或RNA)分子上具有遗传效应的特定核苷酸序列,是细胞内遗传物质的结构和功能单位,可以通过细胞内RNA和蛋白质的合成,决定生物的性状。
2.genome(基因组)是指包含在该生物的DNA(部分病毒是RNA)中的全部遗传信息的总和,也就是单倍体细胞中的全部基因的总和。
人类基因组包括核基因组和线粒体基因组。
3.solitary gene(单一基因)也称单一序列。
是指在一个单倍体基因组中只有一个拷贝的基因。
4.gene family(基因家族)许多基因不是完全单拷贝,属于若干个相似基因的家族,它们进化来源相同,结构、功能相似,称基因家族。
它们可以紧密排列在一起,形成一个基因簇;也可以分散在同一染色体的不同位置,或者存在于不同的染色体上的,各自具有不同的表达调控模式。
5.pseudogene(假基因)是一种畸变基因,其核苷酸序列和有正常功能的基因有很大的同源性;但由于突变而不能表达,因而没有功能。
遗传性代谢缺陷与分子病课件

疾病预防与遗传咨询
遗传风险评估
01
通过家族史、基因检测等手段,评估个体遗传代谢缺陷的风险
,提供针对性的预防建议。
遗传咨询
02
为携带遗传代谢缺陷基因的家庭提供遗传咨询,解释疾病风险
、传递方式等问题,指导家庭生育决策。
新生儿筛查
03
通过大规模新生儿筛查,早期发现潜在的遗传代谢缺陷患者,
进行早期干预和治疗,降低疾病对患儿的影响。
治疗措施
探讨针对该遗传性代谢缺陷的 治疗措施,如饮食控制、酶替
代疗法、基因治疗等。
案例二:一种分子病的综合诊断与治疗
01
02
03
04
案例介绍
详细介绍某种分子病的病例, 包括患者的临床表现、疾病的
遗传基础等。
分子病的发病机制
阐述该分子病的发病机制,包 括相关基因、突变位点、蛋白
质功能异常等。
诊断流程
遗传性代谢缺陷的发病机制涉及基因突变,这些突变可能导致酶活性的降低或丧失,代谢 通路受阻,以及代谢产物在体内的异常累积。这些异常代谢产物可能对机体产生毒性作用 ,引起不同组织和器官的损害。
遗传性代谢缺陷的分类
单基因遗传代谢缺陷
由单一基因突变引起的代谢缺陷,如苯丙酮尿症、囊性纤维化等 。
多基因遗传代谢缺陷
05
典型案例分析
案例一:某种遗传性代谢缺陷的分析
案例介绍
详细介绍某种遗传性代谢缺陷 的病例,包括患者的临床表现
、家族史、遗传方式等。
诊断方法
介绍对该遗传性代谢缺陷的诊 断方法,包括生化检测、基因 检测等手段。
代谢缺陷的生化机制
阐述该遗传性代谢缺陷的生化 机制,如涉及的代谢途径、关 键酶、代谢产物的变化等。
医学遗传学:分子病与先天性代谢缺陷

Hereditary persistence of fetal hemoglobin (HPFH)
6
人类正常血红蛋白的组成
血红蛋白
血红素
血红蛋白单体
7
8
地中海贫血
疾病简介:临床体征、地域分布
分子和细胞病理学机制:致病基因,常见突变, 发病机制
临床处置:诊断、治疗和预防
主要内容:
血红蛋白病 血友病 胶原蛋白病 膜通道蛋白病 结构蛋白病 角蛋白病
氨基酸代谢异常 糖代谢异常 核苷酸代谢异常 脂代谢异常 溶酶体储积症
5
血红蛋白病
• 异常血红蛋白
Abnormal hemoglobin
• 珠蛋白生成障碍性贫血(地中海贫血 ,地贫)
Thalassemia
50%~55 %
1%~3%
发病时间较晚,无自发性出血,在拔牙或外科手术 后异常出血 只有大手术后才发生出血
30
血友病A的致病基因
31
血友病A的常见突变
突变类型 倒位突变 无义突变 插入/缺失突变
中国南方不同高发地
区人群携带率 1 % ~ 23%
地贫的分类:按合成速率降低的珠蛋白类型
14
地中海贫血的致病基因:珠蛋白基因
α-珠蛋白基因突变
缺失型突变 基因簇发生大片段缺失 80%~85%
非缺失型突变 突变导致生成无功能的mRNA 突变导致mRNA加工障碍 突变导致生成不稳定的珠蛋白链
α-珠蛋白基因缺失突变
地贫临床表型差异与相关基因型的关系
2 1
2 1 2
2 1 2 1
2 1 2 1
2 1 2 1
正常 (/ )
静止型 (-/ )
医学遗传学名词解释(生化遗传学)

医学遗传学名词解释(生化遗传学)1、分子病(molecular disease)分子病是指基因突变使蛋白质的分子结构或合成的量异常直接引起机体功能障碍的一类疾病。
包括血红蛋白病、血浆蛋白病、受体病、膜转运蛋白病、结构蛋自缺陷病、免疫球蛋白缺陷病等。
2、先大性代谢缺陷病(inborn errors of metabolism)先天性代谢缺陷也称遗传性酶病,指由于遗传上的原因(通常是基因突变)而造成的酶蛋白质分子结构或数量的异常所引起的疾病。
3、融合基因(fusion gene)融合基因指由两种不同基因的局部片段拼接而成的DNA片段。
4、血友病(hemophilia)血友病是一类遗传性凝血功能障碍的出血性疾病,包括血友病A,血友病B及血友病C。
5、受体病(receptor disease)由于受体蛋白的遗传性缺陷导致的疾病称为受体病。
6、血红蛋白病(hemoglobinopathy)血红蛋白病是由于红蛋白分子合成异常引起的疾病,习惯上分为血红蛋白病和地中海贫血两类。
7、结构蛋白病(structural of protein disease)结构蛋自缺陷病是构成细胞的基本结构和骨架的蛋白的遗传性缺陷引起的疾病,主要包括胶原蛋自病、肌营养不良症等。
8、膜转运蛋白病(membranous transmitted protein disease)由于膜转运蛋白的遗传缺陷导致的疾病称为膜转运蛋白病。
如胱氨酸尿症、囊性纤维样变及先天性葡萄糖、半乳糖吸收不良症等。
9、地中海贫血(tha1assemia )地中海贫血是指由于某种或某些珠蛋白链合成速率降低,造成一些肽链缺乏,另一些肽链相对过多,出现肽链数量的不平衡,而导致的溶血性贫血10、镰状细胞贫血(sick1e cel1 anemia)镰状细胞贫血是因β珠蛋白基因缺陷而引起的一种疾病,呈常染色体隐性遗传。
分子病与先天性代谢缺陷病
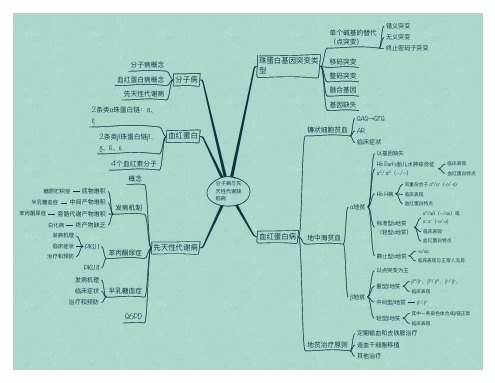
β⁰/β⁺、 β⁰/ β⁰、 β⁺/ β⁺、 临床表现
中间型β地贫 β⁺/ β⁺
轻型β地贫
其中一一条染色色体合成β链正常 临床表现
定期输血血和去铁胺治疗
地贫治疗原则 造血血干干细胞移植
其他治疗
临床表现 血血红蛋白白特点
α地贫
地中海海贫血血
双重杂合子子 α⁰/α⁺(-α/-α)
Hb H病 临床表现
血血红蛋白白特点
标准型α地贫 (轻型α地贫)
α⁰/αA(--/αα)或 α⁺ α⁺(-α/-α) 临床表现 血血红蛋白白特点
静止止型α地贫 以点突变为主
-α/αα 临床表现与正常人人无无异
β地贫
重型β地贫
PKU II 发病机理理 临床症状 治疗和预防
半乳糖血血症
先天性代谢病
G6PD
分子子病与先 天性代谢缺 陷病
珠蛋白白基因突变类 型
单个碱基的替代 (点突变)
移码突变
错义突变 无无义突变 终止止密码子子突变
整码突变
融合基因
基因缺失
血血红蛋白白病
GAG→GTG
镰状细胞贫血血 AR
临床症状
以基因缺失
Hb Bart's胎儿儿水水肿综合征 α⁰/ α⁰(--/--)
分子子病概念 血血红蛋白白病概念
先天白白链:α、 ξ
2条类β珠蛋白白链β、 γ、δ、ε
血血红蛋白白
4个血血红素分子子
概念
糖原贮积症 底物堆积
半乳糖血血症 中间产物堆积 苯丙酮尿尿症 旁路路代谢产物堆积
发病机制
白白化病 终产物缺乏
发病机理理
临床症状 治疗和预防
PKU I
苯丙酮尿尿症
分子病与先天性代谢缺陷病PPT课件

(3)血红蛋白M病:珠蛋白基因突变使血红蛋白和亚铁离子结合的 正常氨基酸被另一个不同氨基酸取代,导致亚铁离子变为铁离子,使 部分铁原子变为稳定的高铁状态,影响了血红素和氧的结合能力,导 致组织缺氧。本病为常染色体显性遗传。
(一) 血红蛋白病
3.地中海贫血 地中海贫血为珠蛋白基因突变导致蛋白肽链合成量异 常而引起的贫血症。 (1)α 地中海贫血:①Hb Bart 胎儿水肿综合征:4个α 珠蛋白基 因全部缺失或缺陷,完全不能合成α 珠蛋白链。胎儿严重缺氧,发育 到8~10个月全身水肿而死亡。②Hb H病:3个α 珠蛋白基因缺失或缺陷, 表现为轻度或中度贫血,患者肝脾大,有轻度或间歇发生黄疸,可发 生继发性感染。③轻型(标准型)α 地中海贫血:2个α 珠蛋白基因缺 失或缺陷。临床轻度溶血性贫血或无症状。④静止型α 地中海贫血:1 个α 珠蛋白基因缺失或缺陷,一般无症状。
(一) 血红蛋白病
(2)β 地中海贫血:①重型β 地中海贫血:患者正常β 链缺乏或合 成量很少,血红细胞中无HbA或HbA含量很少,HbF和HbA2含量增高,α 珠蛋白链过剩而沉积到红细胞膜上,改变了红细胞膜的性能,引发严 重的溶血性贫血。主要临床症状表现为患儿出生几月后溶血反应,并 伴有腹泻、发热、生长发育迟缓、身材矮小、骨髓增生,可出现鼻塌 眼肿、上颌前突、头大额隆等特殊的“地中海贫血面容”。②中间型 β 地中海贫血:临床表现介于轻型和重型之间,中度贫血,脾脏轻或 中度肿大,黄疸可有可无,骨骼改变较轻。③轻型β 地中海贫血:带 有一个正常的β 珠蛋白基因,一般无临床症状或有轻微贫血和脾脏肿 大。
(四) 膜转运载体蛋白病
肝豆状核变性的遗传又称Wilson病,由编码P型铜转运ATP 酶的基因(ATP7Base)突变引起,基因定位于13q14.3,为常 染色体隐性遗传,ATP7Base基因突变多为错义突变或无义 突变。临床表现以神经系统和肝脏受损为主,晚期表现为 痴呆。患者可通过服用青霉胺或锌剂加速清除体内贮积的 铜离子而进行早期治疗,因此本病的早期诊断对于临床治 疗很有意义。
遗传性代谢缺陷与分子病课件

第二节 分子病
分子病是指由于结构基因突变而造成 蛋白质分子结构或合成量异常所引起的疾病。
•遗传性代谢缺陷与分子病
一、血红蛋白病
由血红蛋白分子合成异常引起的疾病称为血红蛋白病。
(a)血红蛋白亚单位 (b) 血红蛋白四聚体 人类正常血红蛋白的结构
•遗传性代谢缺陷与分子病
正常人体血红蛋白
白化病患者
•遗传性代谢缺陷与分子病
尿黑酸尿症
1
发病率
1/250000
2
遗传方式 AR
3
基因定位
3q21-q23
•遗传性代谢缺陷与分子病
4、尿黑酸尿症的发病原理
苯丙氨酸
酪氨酸
尿黑酸(堆积) 尿黑酸氧化酶(缺乏)
乙酰乙酸
CO₂+H₂O
•遗传性代谢缺陷与分子病
5、尿黑酸尿症的临床特征
新生儿期即可由尿排出大量尿黑酸, 新鲜尿的颜色正常,放置空气中则变为棕 色或黑色。成年以后因被氧化的尿黑酸长 期沉积于结缔组织中,致使中廓、巩膜、 鼻、颊等变为褐色或蓝黑色而出现褐黄病, 晚期可累及关节,进展为褐黄病性关节炎。
2
遗传方式 AR
3
基因定位 12q24.1
•遗传性代谢缺陷与分子病
4、苯丙酮尿症的发病原理
苯丙氨酸羟化酶(缺乏)
苯丙氨酸 (堆积)
酪氨酸
酪氨酸酶(活性降低)
苯丙酮酸 (堆积)
黑色素(减少)
苯乳酸(堆积) 苯乳酸(堆积)
•遗传性代谢缺陷与分子病
5、苯丙酮尿症的临床特征
①神经系统症状:如兴奋不安、多 动或嗜睡、萎靡、肌张力增高、腱 反射亢进、惊厥、智能发育落后, 80%有脑电图异常。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
本章重点
• 各类代表性疾病的遗传方式,缺陷基因,致病机制 • 各类代表性疾病的典型临床症状 • 各类代表性疾病的临床处置
2
主要内容
• 血红蛋白病 • 血浆蛋白病 • 胶原蛋白病 • 膜通道蛋白病 • 结构蛋白病 • 角蛋白病
氨基酸代谢异常 糖代谢异常 核苷酸代谢异常 脂代谢异常 溶酶体储积症
地中海贫血的临床体征
➢ 小细胞低色素性溶血型贫血(中、重度) ➢ 黄疸 ➢ 肝脾增大(脾大明显) ➢ 骨髓扩增 ➢ 发育迟缓 ➢ 合并感染
重型地贫
重型地贫
患儿颅骨及面颊部骨骼增大,头颅变大,额部 隆起,颧高,鼻梁塌陷,两眼距离增宽,眼睑
浮肿,形成地中海贫血的特殊面容。
地中海贫血病人带来的家庭和社会负担
血友病A、B、C的共同特征是:凝血活酶的促凝活性降低,凝血 时间延长,轻微外伤后就有终身出血倾向
30
血友病A的临床体征
根据患者FⅧ的促凝成分活性(FⅧ:C,1ml 正常血浆的因子水平定为1U/ml,正常活力 假设为100%,但实际不患血友病的正常个体 的下限为50%)与症状的严重程度,血友病 A可分为4型
• 缺失突变 我国发现5种
β-珠蛋白基因点突变
地贫临床表型差异与相关基因型的关系
临床表型
链 / 非链( or )
轻型 (TT)
基因型
+ / N 0 / N
修饰基因
中间型 (TI)
+ / + 0 / +
基因型
重型 (TM)
0 / + 0 / 0
HbF调 节基因
Hematology Am Soc.2005.
FⅧ基因最常见突变类型为内含子22倒位突变
流行病学
➢ 血友病A最常见,发病人数约占血友病总数的85%,血友病B占 15%~20%
➢ 中国和日本患病率较低(3.0/10万),美国患病率较高(10/10万) ➢ 血友病A的男性发病率约为1/5000,血友病B的男性发病率为1/30
000,女性均罕见,血友病C较为罕见
-地贫: Hb A2 < 2.0%
遗传咨询
珠蛋白生成障碍性贫血遗传方式为常染色体隐性遗传 珠蛋白生成障碍性贫血携带者无临床症状,无需治疗 中间型珠蛋白生成障碍性贫血患者的表型变异度大,很难根据基因
型来准确预测疾病的表型,因此,针对此类患儿的产前诊断应十分 慎重 重型珠蛋白生成障碍性贫血为致死性疾病,建议放弃受累胎儿 夫妇双方中一方为-珠蛋白生成障碍性贫血基因携带者,另一方为珠蛋白生成障碍性贫血基因携带者时,不需要进行产前诊断 少量-珠蛋白生成障碍性贫血家系中的遗传规律为常染色体显性遗 传
25
地贫预防策略:婚检、产检
2002-2010年广西婚检率和重型地贫发生率情况比较
婚检率(%)
产前诊断干预使高发区地中海贫血患儿出生率下降
Prenat Diagn,2005;中华医学遗传学杂志,2008;三地实施现场统计资料.
南方医科大学南方医院儿科移植经验
血友病
• 疾病简介:临床体征、流行病学 • 分子和细胞病理学机制:致病基因,常见突变,
2 1
2 1 2
2 1 2 1
2 1 2 1
2 1 2 1
正常 (/ )
静止型 (-/ )
标准型 (-/ -) (--/ )
Hb H 病 Hb Barts水肿胎
(- -/-)
(- -/- - )
β-珠蛋白基因突变
• 点突变 至少300种 启动子突变、3’UTR突变、异常剪接突变 起始密码突变、无义突变、移码突变 显性突变、小缺失
3
分子病和先天性代谢缺陷
• 分子病(molecular disease)
基因突变导致编码的蛋白质发生结构或数量上的变化,从 而导致机体出现病理变化及功能障碍,由此引起的疾病
• 先天性代谢缺陷(inborn errors of metabolism )
编码酶蛋白质的基因突变引起酶合成障碍,使机体的代谢 过程不能正常进行,最终ห้องสมุดไป่ตู้致疾病
地贫的分类:按合成速率降低的珠蛋白类型
15
地中海贫血的致病基因:珠蛋白基因
α-珠蛋白基因突变
• 缺失型突变 基因簇发生大片段缺失 80%~85%
• 非缺失型突变 突变导致生成无功能的mRNA 突变导致mRNA加工障碍 突变导致生成不稳定的珠蛋白链
α-珠蛋白基因缺失突变
地贫临床表型差异与相关基因型的关系
Hereditary persistence of fetal hemoglobin (HPFH)
6
人类正常血红蛋白的组成
血红蛋白
血红素
血红蛋白单体
7
异常血红蛋白
8
地中海贫血
• 疾病简介:临床体征、地域分布 • 分子和细胞病理学机制:致病基因,常见突变,
发病机制 • 临床处置:诊断、治疗和预防
重型α地贫
中晚孕期死胎,孕妇并发重度妊高症, 每例人均误工费和治疗费5万元以上。
重型β地贫 每年须花5万元以上治疗费来维持生命。
中间型α和β地贫
终生贫血,大部分没有正常劳动力,每 例人均年治疗费和生活费5万元以上。
地中海贫血的地域分布
Thalassemia
中国南方不同高发地
区人群携带率 1 % ~ 23%
4
Garrod(1908)提出先天性代谢缺陷 的概念
Pauling(1949)提出分子病的概念
遗传方式大多数为AR ,少数为XR, 极少数为AD
血红蛋白病
• 异常血红蛋白
Abnormal hemoglobin
• 珠蛋白生成障碍性贫血(地中海贫血 ,地贫)
Thalassemia
• 遗传性持续性胎儿血红蛋白持续存在综合征
发病机制 • 临床处置:诊断、治疗和预防
血友病
疾病介绍
➢ 属血浆蛋白病,是一组遗传性凝血因子缺乏的出血性疾病 ➢ 分为血友病A、血友病B、血友病C和血管性假血友病 ➢ 血友病A与血友病B常见,均呈X连锁隐性遗传,分别由FⅧ与FⅨ基
因缺陷导致 ➢ 血友病A是由于FⅧ基因突变导致正常凝血因子Ⅷ活性降低导致的,
地贫的临床遗传学
遗传检测 血液学表型诊断 临床表型诊断 基因型诊断 人群筛查和产前诊断
治疗 定期的输血和铁螯合剂去铁 干细胞移植
23
血液学诊断
α-地贫: MCV < 80 fL, MCH< 27 pg 而且 Hb A2 < 2.5%; (对静止型地贫不适用)
β-地贫: MCV < 80 fL, MCH < 27 pg 而且 Hb A2 > 3.5%;