金属酞菁的合成及表征
酞菁铁Ⅱ的制备及表征

酞菁铁(Ⅱ)的制备及表征武汉大学化学与分子科学学院王小尚 200331050033摘要:通过制备Fe(OH)2·4H2O制备酞菁铁(Ⅱ), 并对产品进行纯化,通过紫外及红外的方法分析确定其组成关键字:酞菁铁(Ⅱ);制备;纯化;红外;紫外分光法1.前言酞菁类化合物可以看成四氮杂卟啉的衍生物,具有D2h点群对称性。
其在染料工业和光电功能材料等方面获得了巨大的应用,并具有电致变色效应,在室温下有很好的液晶相,也在催化剂,抗辐射剂等方面也有重要作用。
酞菁类化合物的合成一般采用Linstead合成方法,其提纯比较困难。
反应产物中含有大量的杂质,包括原料和一些其他高分子聚合物,常用的提纯方法有微热丙酮索氏萃取除杂,真空升华,浓H2SO4再沉淀或色谱柱提纯。
合成酞菁铁的前体有:邻苯二甲酸,邻苯二甲酸酐,邻苯二甲氰,邻苯二甲酸氨酯等。
本实验以邻苯二甲酸酐,Fe(OH)2·4H2O(自制),尿素为原料,以(NH4)2MoO4为催化剂,采用固相熔融法合成FePc,用真空升华法提纯产物,纯产物经元素分析,红外及紫外可见光谱表征。
2.实验部分2.1试剂及仪器:1.试剂还原铁粉,6mol/L盐酸,邻苯二甲酸酐,尿素,乙醇,10%氢氧化钠,酸铵,浓硫酸2.仪器减压过滤装置,旋转蒸发仪,真空干燥器,量筒(50mL),三口瓶(250mL,100mL),滤纸,烧杯(250mL),24#圆底烧瓶(100mL),24#空气冷凝管,24#磨口弯头,24#磨口塞,油泵,19#导气管,橡皮管,电热套(250mL), 研钵,温度计(3000C),长玻棒,容量瓶(50mL)表面皿,牛角勺,天平,氮气钢瓶,管式电炉,旋子流量计,石英管,烘箱,小瓷舟,UV-Vis 分光光度计,红外光谱仪。
2.2实验步骤:1. FeCl2·4H2O的制备称取5.67g还原铁粉放入100 mL的三口烧瓶中,并向其中加入30 mL6mol/L的盐酸溶液,缓缓通入氮气至液面下,烧瓶上的一个瓶口用导气管将逸出气体(包括反应的生的H2和为了防止氧化而通入的N2以及少量HCl气体)通经过安全瓶(防倒吸)导入稀碱溶液(中和逸出的少量HCl气体)。
金属酞菁的合成及表征
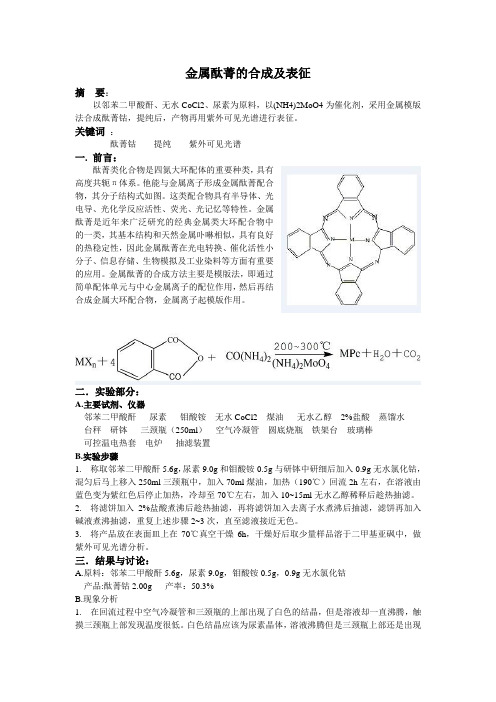
金属酞菁的合成及表征摘要:以邻苯二甲酸酐、无水CoCl2、尿素为原料,以(NH4)2MoO4为催化剂,采用金属模版法合成酞菁钴,提纯后,产物再用紫外可见光谱进行表征。
关键词:酞菁钴提纯紫外可见光谱一. 前言:酞菁类化合物是四氮大环配体的重要种类,具有高度共轭π体系。
他能与金属离子形成金属酞菁配合物,其分子结构式如图。
这类配合物具有半导体、光电导、光化学反应活性、荧光、光记忆等特性。
金属酞菁是近年来广泛研究的经典金属类大环配合物中的一类,其基本结构和天然金属卟啉相似,具有良好的热稳定性,因此金属酞菁在光电转换、催化活性小分子、信息存储、生物模拟及工业染料等方面有重要的应用。
金属酞菁的合成方法主要是模版法,即通过简单配体单元与中心金属离子的配位作用,然后再结合成金属大环配合物,金属离子起模版作用。
二.实验部分:A.主要试剂、仪器邻苯二甲酸酐尿素钼酸铵无水CoCl2 煤油无水乙醇2%盐酸蒸馏水台秤研钵三颈瓶(250ml)空气冷凝管圆底烧瓶铁架台玻璃棒可控温电热套电炉抽滤装置B.实验步骤1. 称取邻苯二甲酸酐5.6g,尿素9.0g和钼酸铵0.5g与研钵中研细后加入0.9g无水氯化钴,混匀后马上移入250ml三颈瓶中,加入70ml煤油,加热(190℃)回流2h左右,在溶液由蓝色变为紫红色后停止加热,冷却至70℃左右,加入10~15ml无水乙醇稀释后趁热抽滤。
2. 将滤饼加入2%盐酸煮沸后趁热抽滤,再将滤饼加入去离子水煮沸后抽滤,滤饼再加入碱液煮沸抽滤,重复上述步骤2~3次,直至滤液接近无色。
3. 将产品放在表面皿上在70℃真空干燥6h,干燥好后取少量样品溶于二甲基亚砜中,做紫外可见光谱分析。
三.结果与讨论:A.原料:邻苯二甲酸酐5.6g,尿素9.0g,钼酸铵0.5g,0.9g无水氯化钴产品:酞菁钴2.00g 产率:50.3%B.现象分析1. 在回流过程中空气冷凝管和三颈瓶的上部出现了白色的结晶,但是溶液却一直沸腾,触摸三颈瓶上部发现温度很低。
酞菁的制备和纯化

钼酸铵4邻苯二甲酸酐+4尿素+M2+MPc+H2O+CO22.1.2 金属酞菁的制备和纯化金属酞菁(MPc)按如下模板反应制备:(M=Mn,Cu,Ni,Co)()对于不同的中心离子M2+,具体制备方法也不同。
(1)酞菁锰(MnPc)的制备和纯化苯酐5.92g尿素9.01g锰1.69g钼酸铵2.47*10-3 g加入量:苯酐5.92 (0.04 mol),尿素9.01(0.15mol),钼酸铵2.47*10-3(2*10-6mol),锰1.69(0.01mol)。
一定量的苯酐和尿素置于250ml三颈烧瓶中,加入千分之二的钼酸铵作催化剂,再加入150ml二甲苯作溶剂。
加热至120℃使固体完全溶解,趁热加入硫酸锰。
升温至140℃下回流,20min后溶液变混浊,升温至150℃回流1h,溶液变清,底部有浅黄色沉淀。
倒出二甲苯,160o C下恒温3h蒸出溶剂。
粗产品用6M HCl 浸泡12h,在烧杯中静置后,倒掉上层清液体,反复用蒸馏水洗涤,静置,直至倒出液体为无色且中性。
再用丙酮浸泡,静置,洗至倒出的上层清液为无色。
再用1mol/L的NaOH溶液浸泡(时间?),静置,倒掉上层清夜,再用蒸馏水洗至倒出液为无色且为中性。
在100℃下干燥12h,即得MnPc。
(2)酞菁铜(CuPc)的制备和纯化在250ml三颈烧瓶中将苯酐、尿素和氯化铜按4:4:1的摩尔比混合,再加入千分之二的钼酸铵作催化剂,加入150ml二甲苯作溶剂。
加热,在160℃下回流,20min后溶液变混浊,在此温度下继续回流0.5h,溶液变清,并呈浅蓝色,烧瓶底部有蓝色沉淀。
在200℃下继续回流4h,蒸出溶剂。
粗产品置于6N HCl 中,浸泡12h,过滤,用蒸馏水将蓝色沉淀洗至滤出液为无色,再用丙酮洗至滤出液为无色。
在120℃下干燥12h,即得CuPc。
(3)酞菁镍(NiPc)的制备和纯化苯酐、尿素和硫酸镍配料的摩尔比为4:4:1,先将苯酐、尿素置于250ml三颈烧瓶中,加入千分之二的钼酸铵作催化剂,再加入150ml二甲苯作溶剂。
新型四取代酞菁及金属酞菁的合成与表征

山西大学学报(自然科学版)30(4):493~497,2007Journal of Shanx i U niv ersit y(Nat.Sci.Ed.) 文章编号:0253-2395(2007)04-493-05新型四取代酞菁及金属酞菁的合成与表征夏道成1,马春雨1,程传辉2,丛方地3,于书坤2,常玉春1,2,李万程2,于海丰3,杜国同1,2*(1.大连理工大学三束材料改性国家重点实验室,物理与光电工程学院,辽宁大连116023;2.吉林大学电子科学与工程学院,吉林长春130012;3.东北师范大学化学学院,吉林长春130024)摘 要:以4-硝基邻苯二腈(1)、3-硝基邻苯二腈(4)和2-异丙基-5-甲基苯酚(2)为起始化合物,在氢氧化锂的作用下,经亲核取代反应生成了相应的邻苯二腈衍生物(3)和(5).以化合物(3)和(5)为前体,在催化剂1,8-二氮杂二环[5,4,0]十一碳-7-烯(DBU)的作用下发生环合四聚反应,得到了具有优良溶解性的四取代酞菁,铟氯酞菁和钛氧酞菁6a-6c,7a-7c.并分别用氢核磁、质谱、紫外可见光谱和元素分析确认了这些化合物的结构.关键词:酞菁;3-硝基邻苯二腈;4-硝基邻苯二腈;合成中图分类号:O614 文献标识码:A0 引言酞菁作为一种优异的功能材料,在众多领域被广泛应用,包括染料[1]、化学传感器[2]、非线性光学[3]、癌症的光动力学治疗[4,5]等等.上述应用都要求酞菁须具备优良的溶解性.一般来说,非取代酞菁和非取代金属酞菁只溶于浓硫酸或浓磷酸,而微溶甚至不溶于有机溶剂.提高酞菁在有机溶剂中溶解性的公认方法是对其进行衍生化,通常是在酞菁分子外围苯环上引入可溶性官能团,以提高其溶解性.氨基[6]、烷氧基[7]、磺酰基[8]等都可作为酞菁环上的取代基.这种衍生化通常有两种方式:一种是以酞菁为母体进行的;而另一种是先将邻苯二腈等前体衍生化,再进行环和四聚反应生成酞菁.后者易提纯、产率高,为大多数酞菁合成所采用.本文采用的也是第二种方法,以3-硝基邻苯二腈或4-硝基邻苯二腈为前体首次合成溶解性很好的四取代酞菁,铟氯酞菁和钛氧酞菁6a-6c,7a-7c,见图1(P494).1 实验1.1 仪器和药品3-硝基邻苯二腈(1)和4-硝基邻苯二腈(4)(分析纯,日本东京化成);DBU(分析纯,日本东京化成).LDI-1700激光解吸飞行时间质谱仪(美国);Q USTAR-T OF质谱仪(美国);V ar ian Unity-500型核磁共振仪(美国);Cary-500UV-Vis-NIR光谱仪(美国);Flash EA1112元素分析仪(意大利).1.2 化合物的合成1.2.1 3-(2-异丙基-5-甲基苯氧基)邻苯二腈(3)1.73g(10mmo l)3-硝基邻苯二腈和1.5g(10mmo l)2-异丙基-5-甲基苯酚混合溶解在30mL干燥的DM SO中,然后加入0.42g(10m mol)LiOH・H2O,30℃反应24h.反应完毕后,将反应液倒入400mL质量浓度(下同)为10%的NaCl水溶液中,析出的固体常温干燥后柱层析分离提纯(流动相:石油醚/乙醚=6∶1),得2.23g无色晶体3-(2-异丙基-5-甲基苯氧基)邻苯二腈,产率81%.收稿日期:2007-06-24 基金项目:国家自然科学基金(60307002;20472014) 作者简介:夏道成(1976-),男,博士研究生,从事新型酞菁合成及其光电特性研究.E-mail:x iadaocheng@y .杜国同(1945-),男,教授,博士生导师,从事微电子学与固体电子学研究.E-ma il:dug t@.图1 6a -6c 和7a -7c 的合成路线图F ig.1 T he synthetic r out o f 6a -6c and 7a -7c 1H NM R (500M Hz,CDCl 3): =7.543(dd,1H,J=8Hz,ArH),7.425(d,1H,J=8Hz,ArH ),7.286(d,1H,J=8Hz,A rH),7.091(d,1H ,J=9Hz,ArH ),6.971(d,1H,J=9Hz,Ar H),6.769(s,1H ,ArH ),3.023(m ,1H ,CH ),2.320(s ,3H ,CH 3),1.182(d ,6H ,2CH 3);MS (QU STAR -TOF ):m /z =299.0597(an isotopiccluster peaking )(Calcd .299.1160[M +Na +]);IR (KBr ):=2229cm -1(m ,CN ),1242(w ,C -O -C );UV /VIS(CHCl 3): max =321nm.1.2.2 4-(2-异丙基-5-甲基苯氧基)邻苯二腈(5)1.73g (10mmo l )4-硝基邻苯二腈和1.5g (10mmo l )2-异丙基-5-甲基苯酚混合溶解在30mL 干燥的DM SO 中,然后加入0.42g (10m mol)LiOH ・H 2O,30℃反应24h.反应完毕后,将反应液倒入400m L 质量浓度为10%的NaCl 水溶液中,析出的固体常温干燥后柱层析(SiO 2)分离提纯(流动相:石油醚/乙醚=9∶1),得2.29g 无色晶体4-(2-异丙基-5-甲基苯氧基)邻苯二腈,产率83%.UV -v is (CHCl 3): max :261nm,297nm,306nm;1H NM R(CDCl 3,500M Hz) :7.708(d,J =8.5Hz,1H ),7.295(d,J=8.5Hz,1H ),7.219(s,1H),7.189(d,J=8Hz,1H),7.109(d,J=8Hz,1H ),6.745(s,1H ),2.958(m ,1H ),2.331(s ,3H ),1.154(d ,6H );IR (KBr ) :2229cm -1(CN ),1246cm -1(C -O -C );HRM S calcd for C 18H 16N 2O 2Na 299.1160,found 298.9497.1.2.3 6a 和7a 的合成将4-(2-异丙基-5-甲基苯氧基)[9]邻苯二腈(1.16g ,4.2m mol )(3)或3-(2-异丙基-5-甲基苯氧基)[9]邻苯二腈(5)(1.16g ,4.2mmol )直接在DBU 的催化下成环.溶剂为正戊醇,反应温度为135℃.6a 为蓝绿色粉末,硅胶柱层析流动相为二氯甲烷,产率61%;7a 为绿色粉末,流动相为乙醚/石油醚=2∶9,产率46%.6a :U V/Vis(chloro naphthalene, max /nm )343,612,646,676,710;1H NMR(500M Hz,CDCl 3) :2.037(s ,2H ,NH ),1.428~1.479(m ,24H ,CH 3),2.379~2.462(t ,12H ,CH 3),3.586~3.599(m ,4H ,CH ),7.182(t,8H,Ar H),7.479~7.502(m,4H ,ArH ),7.681(s,4H,ArH ),8.636(s,4H,ArH ),9.019(s,4H,ArH );M S:m/z=1107.7(M +H +);元素分析,C 72N 8H 66O 4,计算值(%):C 78.09,H 6.01,N 10.12;实验值(%):C 78.01,H 6.30,N 10.24.7a :UV/Vis (chlor onaphthalene, max /nm )344,631,665,699,729;1HNMR(500MH z,CDCl 3) :2.016(s,2H,NH ),1.449~1.498(m ,24H,CH 3),2.395(t,12H ,CH 3),3.665~3.757(m,4H,CH ),6.994~7.076(m ,4H ,A rH ),7.138~7.230(m ,4H ,ArH ),7.420~7.509(m ,4H ,ArH ),7.965~8.036(m ,4H ,ArH ),8.888~8.990(m,4H ,ArH),9.156~9.236(m ,4H ,ArH );M S:m/z=1107.6(M +H +);元素分析,C 72N 8H 66O 4,计算值(%):C 78.09,H 6.01,N 10.12;实验值(%):C 78.31,H 6.00,N 9.90.1.2.4 6b 和7b 的合成将4-(2-异丙基-5-甲基苯氧基)[9]邻苯二腈(3)(1.16g ,4.2mm ol)或3-(2-异丙基-5-甲基苯氧基)[9]邻苯494山西大学学报(自然科学版) 30(4) 2007 二腈(5)(1.16g ,4.2m mol)和InCl 3・2H 2O (0.3084g ,1.05mmol)搅拌溶解于8mL 的喹啉中,加入6滴DBU ,在反应体系中充入氮气保护,在180℃反应12h .反应溶液冷却到室温后,加入甲醇析出固体,抽滤,将所得固体用甲醇在索氏提取器中提取24h.然后用硅胶柱层析分离,6b 为绿色粉末:流动相为石油醚/乙醚=2∶1,产率61%;7b 为绿色粉末,流动相为石油醚/乙醚=3∶2,产率44%.6b :UV /Vis (chlo ronaphthalene , max /nm ):359,638,710;1HNM R (500MHz ,CDCl 3) :1.352~1.420(m ,24H ,CH 3),2.420(t ,12H ,CH 3),3.489~3.528(m ,4H ,CH ),7.107~7.192(m ,8H ,ArH ),7.442~7.476(m ,4H,ArH ),7.789~7.843(m ,4H,ArH ),8.789~8.860(m ,4H,ArH ),9.262~9.317(m ,4H,ArH ).M S:m /z=1255.5(M +H +);元素分析,分子式:C 72N 8H 64O 4InCl,计算值(%):C 68.87,H 5.14,N8.92;实验值(%):C 68.94,H 5.21,N 8.78.7b :UV/Vis (chloronaphthalene, max /nm )359,661,738;1HNM R(500M Hz,CDCl 3) :1.478~1.560(m,24H ,CH 3),2.357~2.393(m ,12H,CH 3),3.708~3.782(m,4H,CH),6.992~7.054(m ,4H,ArH ),7.162~7.240(m ,4H ,Ar H ),7.476~7.551(m ,4H ,ArH ),7.989~8.068(m ,4H ,ArH ),8.965~9.065(m ,4H ,ArH ),9.220~9.312(m ,4H ,A rH );M S :m /z =1256.2(M ,isotopic cluster );元素分析,C 72N 8H 64O 4InCl,计算值(%):C 68.87,H 5.14,N 8.92.实验值(%):C 68.63,H 5.26,N 8.81.1.2.5 6c 和7c 的合成步骤和6b ,7b 的制法基本相同,InCl 3・2H 2O 改为T i(OBu)4,溶剂为正戊醇,反应温度为155℃.硅胶柱层析时先用石油醚/二氯甲烷= 1.5∶1的流动相除去杂质,再用甲醇/二氯甲烷=1∶40淋洗,得到6c ,蓝绿色粉末,产率45%;先用石油醚/二氯甲烷=1∶1的流动相除去杂质,再用甲醇/二氯甲烷=1∶30淋洗,得到7c ,蓝绿色粉末,产率35%.6c :UV /Vis (chloro naphthalene, max /nm )349,397,639,712;1HNM R(500MH z,CDCl 3) :1.36~1.444(m ,24H ,CH 3),2.421~2.442(m ,12H ,CH 3),3.538~3.586(m ,4H ,CH ),7.177~7.223(m ,8H ,ArH ),7.453~7.483(m ,4H ,ArH ),7.778~7.815(t ,4H ,ArH ),8.731(s ,4H ,Ar H ),9.065~9.170(m ,4H ,ArH );M S:m/z=1168.6(M +H +);元素分析,C 72N 8H 64O 5T i,计算值(%):C 73.96,H 5.52,N 9.58;实验值(%):C 73.83,H 5.40,N 9.86.7c :U V /Vis (chloro naphthalene , max /nm )350,428,661,738;1HNM R (500M Hz ,CDCl 3) :1.499~1.546(m,24H ,CH 3),2.372~2.418(m,12H,CH 3),3.725~3.787(m,4H ,CH ),7.026~7.079(m ,4H,ArH),7.182~7.244(m,4H,ArH),7.521~7.59(m,4H ,ArH),8.037~8.113(m,4H ,ArH),9.057~9.167(m ,4H ,ArH ),9.310~9.391(m ,4H ,ArH );M S :m /z =1168.2(M ,isotopic cluster );元素分析,C 72N 8H 64O 5T i ,计算值(%):C 73.96,H 5.52,N 9.58;实验值(%):C 74.01,H5.40,N 9.67.2 结果与讨论2.1 合成和以往的合成酞菁方法不同[10],酞菁6a 和7a 采用在DBU 的催化下直接缩合成环.与金属锂催化[10]成环的方法相比,此法的优势在于步骤比较简便,而产率亦较为理想.金属酞菁6b 和7b 的制备须要在180℃左右才可完成,原因可能是金属铟必须在较高的温度下才可以和酞菁环生成配合物.前体(3)和(5)在制备6c 和7c 的过程中会有一定量的6a 和7a 生成,质谱可以证明6a 和7a 的存在.这可能是由于T i(OBu)4的性质决定的.2.2 紫外分析图2(P496)和图3(P496)给出了酞菁和金属酞菁6、7的紫外可见吸收光谱.从图中可以清晰的看到酞菁和金属酞菁的特征吸收带:B 带(通常在300nm ~400nm 之间)和Q 带(通常的620nm ~800nm 左右).Q 带的存在也表明了酞菁环的形成,和金属酞菁的Q 带相比,酞菁的Q 带分成两部分,这主要是由酞菁分子的对称性决定的.从图中也可看出,在酞菁环!位取代会比∀位取代对Q 带的红移贡献大.以6c 和7c 为例,!位取代的7c 会比∀位取代6c 红移约26nm.Q 带的存在也证明了酞菁环的形成.495 夏道成等:新型四取代酞菁及金属酞菁的合成与表征图2 6a 和7a 的紫外谱图Fig.2 U V /V is spectra o f Pc 6a and7a 图3 6b ,7b ,6c 和7c 的紫外谱图Fig.3 U V /Vis spectra of Pc 6b ,7b ,6c and 7c2.3 质谱分析图4和图5给出了金属酞菁7c 和7b 的质谱.三个强度最高的数值1169.2,1255.0和1277.0分别对应着[M 2c +H +],[M 2b +H +]和[M 2b +Na +],从图中也可以清晰的看到两种化合物的同位素峰.质谱的实验值与理论值吻合的很好.酞菁和金属酞菁6a -6c ,7a -7c 的氢核磁谱比较复杂,难于归属.这主要是由于它们的聚合效应和同分异构体造成的,但是,氢核磁谱中的信号与它们结构是吻合的.图4 7c 的质谱图F ig .4 M A L DI -T OF M S o f Pc 7c 图5 7b 的质谱图F ig .5 M A L DI -T OF M S o f Pc 7b2.4 结构分析酞菁6a ,7a 的结构式经Chem 3D U ltra 8.0软件进行分子势能最小化计算,得出了如图6,图7所示的结构图,这将有助我们更清楚了解它们的结构.图6 6a 的结构图F ig .6 T he st ructure o f 6a 图7 7a 的结构图F ig .7 T he structur e of 7a3 结论综上所述,我们合成了酞菁和金属酞菁6a -6c ,7a -7c ,并通过了氢核磁谱、质谱、元素分析及紫外可见光谱表征了它们的分子结构.预期这些化合物作为电致发光材料具有进一步开发及应用前景.496山西大学学报(自然科学版) 30(4) 2007 参考文献:[1] SHEN Y J .T he Syntheses and A pplicatio n of P ht halo cy anines [M ].Beijing :Chemica l Industr y P r ess ,2000:6-7.[2] L EE Y L ,HSI AO C Y ,CHA NG C H,et al .Effects of sensing temperatur e o n t he G as Sensing Pr oper ties of CopperP hthalo cyanine and Co pper T etr a-ter t-buty l P hthalo cyanine Films[J].S ensor s and A ctuators B ,2003,94:169-175.[3] SA ST RE A ,BEL EN D R,T OR RES T.Synthesis o f N ovel U nsymmetr ically Substit ut ed Push-P ull P hthalocyanines[J].J Org Chem ,1996,61:8591-8597.[4] A L I H,L IER J E V A N.M et al Co mplex es as P hoto -and R adiosensitizer s[J].Chem Rev ,1999,99:2379-2450.[5] F A U ST R.T he M o dular Appro ach t o A cetylenic Phthalocy anines and Phthalocyanine A nalo g ues[J].E ur J Org Chem ,2001:2797-2803.[6] CO N G F D ,N IN G B ,DU X G ,et al .F acile Synthesis ,Char acterizat ion and Pr operty Compar isons of T etra aminometa ll -o phthy lo cyanines w ith and w it ho ut Intr amolecular Hy dr og en Bo nds[J].Dy es and P igments ,2005,66:149-154.[7] R AG ER C,SCHM ID G,HA N A CK M .Influence of Substituents,R eact ion Conditions and Centr al M etals o n the IsomerD istributions of 1(4)-T etr asubstit ut ed P ht ha lo cy anines [J ].ChemE ur J ,1999,5(1):280-288.[8] JIA N G X E ,G U O L P ,DU X G .Elect ro chemist ry a nd Electr ocatalysis o f Binuclear Co balt P hthalo cyanine -hex asulfo na -t e-surfactant F ilm M o dified Electro de [J].T alanta ,2003,61:247-256.[9] M A C Y ,T IA N D L ,HOU X K ,et al .Sy nthesis and Char acter izatio n o f Sev era l Soluble T et rapheno x y-substituted Co p-per and Zinc P ht halo cy anines [J ].Sy nthesis ,2005,5:0741-0748.[10] CHO I C F ,T SA N G P T ,HU A N G J D ,et al .Synthesis and in V itr o Pho todynamic A ctiv ity o f new Hex adeca-carbo xy p-hthalo cyanines[J].Chem Commun ,2004:2236-2237.Synthesis and Characterization of Novel Tetra -substitutedPhthalocyanines and MetallophthalocyaninesXIA Dao -cheng 1,M A Chun-yu 1,CH ENG Chuan-hui 2,CONG Fang-di 3,YU Shu-kun 2,CHANG Yu-chun 1,2,LI Wan-cheng 2,YU Hai-feng 3,DU Guo-tong 1,2(1.S tate K ey L abor atory of M aterials M od if ication by L aser ,I on and Electr on B eams ,School of Phy sics andOp toelectr onic T echnology ,Dalian U niv er sity of T echnology ,Dalian 116023,China ;2.State K ey L aborator y of I ntegr atedOp toelectr onics ,College of Electr onic Science and Eng ineer ing ,J ilin U niv er sity ,Changchun 130012,China ;3.Faculty of Chemistry ,N ortheast N or mal U niver sity ,Changchun 130024,China )Abstract :Phthalo cyanines (Pcs ),Chloroindium phthalocy anines (ClInPcs )and ox otianium phthalocyanines (OTiPcs)6a -6c ,7a -7c w er e gener ated by cy clotetramerisation of phthalonitrile derivativ es (3)and (5)in the presence of 1,8-diazabicy clo [5.4.0]undec-7-ene (DBU )as the catalyst.T aking 4-nitrophthalonitrile(1)or 3-nitrophthalonitr ile (4)w ith 2-isopropyl -5-m ethylphenol (2)as the starting materials ,the co rresponding phthalonitr ile derivativ es (3)and (5)w ere prepared .All the compounds w ere characterized by 1H NM R,M S,U V/Vis and elem ental analysis,w hich w ere co nsistent w ith the pro posed structur es.Key words :Sy nthesis;phthalocyanine;3-nitrophthalonitrile;4-nitrophthalonitrile 497 夏道成等:新型四取代酞菁及金属酞菁的合成与表征。
金属酞菁的合成及应用研究的开题报告

金属酞菁的合成及应用研究的开题报告
题目:金属酞菁的合成及应用研究
一、选题背景及意义
金属酞菁是一种重要的有机金属配合物,具有良好的光学和电学性质,在光电领域、光敏电子器件、有机场效应晶体管等领域都有广泛的
应用。
因此,研究其合成及应用具有重要的实际意义。
二、研究目的及内容
本研究的目的是通过合成不同金属酞菁(例如,铜酞菁、钴酞菁、
锰酞菁等),探究其在光电器件中的应用,同时通过对其光电性能进行
表征和分析,探究金属酞菁在光电领域的应用前景。
具体研究内容如下:
1. 制备不同金属酞菁
在实验室中制备不同金属酞菁,如铜酞菁、钴酞菁、锰酞菁等,并
对其微观结构和晶体结构进行表征和分析。
2. 测量金属酞菁的光电性能
使用光电性能测试仪器对金属酞菁的光电性能进行测量,如吸收光谱、发光光谱、电化学行为等等。
3. 探究金属酞菁在光电器件中的应用
通过实验探究不同金属酞菁在光电器件中的应用,如有机场效应晶
体管、光敏电子器件等,研究其在光电功能材料方面的应用前景。
三、研究方法及步骤
1. 液相法合成金属酞菁
2. 对金属酞菁进行晶体结构表征
3. 测量金属酞菁的光电性能
4. 探究金属酞菁在光电器件中的应用
四、预期成果及意义
本研究预期能够合成多种金属酞菁,并对其在光电器件中的应用进行探究,为相关技术的发展做出一定的贡献,同时推动金属酞菁在材料学、物理学和化学等领域的广泛应用。
金属酞菁

实验六金属酞菁配合物的合成及光谱性质研究一实验目的(1)通过合成酞菁金属配合物,掌握这类大环配合物的一般合成方法,了解金属模板反应在无机合成中应用。
(2)进一步熟练掌握配合物合成中的常规操作方法和技能。
二实验原理金属酞菁的合成自由酞菁(H2Pc)的分子结构见图1(a)。
它是四氮大环配体的重要种类,具有高度共轭π体系。
它能与金属离子形成金属酞菁配合物(MPc),其分子结构式如图1(b)。
这类配合物具有半导体、光电导、光化学反应活性、荧光、光存储等特性。
金属酞菁是近年来广泛研究的经典金属大环配合物中的一类,其基本结构和天然金属卟啉相似,且具有良好的热稳定性和化学稳定性,因此金属酞菁在光电转换、催化活化小分子、信息储存、气敏传感器、生物模拟及工业染料等方面有重要的应用。
N N HNNNHNN NNNNNNNN NMM = Cu,Co,Ni,Zn,Pb,Pda b图1 酞菁配合物的结构示意图金属酞菁的合成一般有以下两种方法:①通过金属模板反应来合成,即通过简单配体单元与中心金属离子的配位作用,然后再结合形成金属大环配合物。
这里的金属离子起着一种模板作用;②与配合物的经典合成方法相似,即先采用有机合成的方法制得并分离出自由的有机大环配体,然后再与金属离子配位,合成得到金属大环配合物。
其中模板反应是主要的合成方法。
金属酞菁配合物的合成的方法主要有以下几种途径(以2价金属M为例)。
(1) 中心金属的置换MX + LiPcMPc + 2LiX(2) 以邻苯二甲腈为原料MX n +CNCN4MPc℃300溶 剂(3) 以邻苯二甲酸酐、尿素为原料ΔMX n +CoCo4MPc℃300O+ CO(NH 2)2200 ~424(4) 以2-氰基苯甲酸胺为原料M +CNCONH 24MPc + H 2O℃250Δ本实验按反应(2)制备金属酞菁,原料为金属盐、邻苯二甲腈,催化剂为1,8-二氮杂双环[5,4,0]十一-7-烯(DBU)。
金属酞菁的合成实验报告

金属酞菁的合成实验报告
实验目的:通过合成过程了解金属酞菁分子结构和性质,掌握实验中各种试剂的使用
方法和实验操作技能,以及掌握操作规范与安全常识。
实验原理:金属酞菁是由酞菁分子与金属离子配位形成的配合物,其中金属离子常见
的有Co、Cu、Fe等。
合成金属酞菁一般采用先制备酞菁钠,将其与金属盐在适当反应条件下反应即可得到金属酞菁。
实验步骤:
1. 酞菁钠的制备
取称量好的酞菁(0.2g)放入三颈瓶,加入甲苯(50ml)和氢氧化钠(1g),用磁力
搅拌器搅拌至溶解,然后在75℃下进行热反应2小时,反应完毕后离心,将上层透明的溶液过滤,过滤液收集并去掉甲苯,冷却后得到暗绿色的酞菁钠晶体,为下一步反应的原料。
(反应方程式为:H2C2N4M + NaOH → Na2H2C2N4 + H2O + M(OH)2)
结果分析:金属酞菁制备成功,样品为暗绿色结晶,红外光谱图中有明显的吸收峰,
符合金属酞菁的典型结构;元素分析结果为C:64.17%、H:2.79%、N:18.84%、Cu:
6.87%,符合理论值,说明金属酞菁合成得到。
结论:通过实验合成了金属酞菁,得到了暗绿色结晶的样品,且经元素分析、红外光
谱验证得到的样品符合金属酞菁的理论结构,合成过程成功。
不同取代基金属酞菁的合成表征及光谱性质研究

不同取代基金属酞菁的合成表征及光谱性质研究相比非取代酞菁,取代金属酞菁不仅在有机溶剂中具有更好的溶解性,而且由于这类物质结构中电子共轭大环体系的存在,也使得它们在光、电、磁领域的研究中有着广泛的应用前景。
因此,设计并合成具有优良电致发光性能和光伏性能的金属酞菁化合物越来越成为研究的热点。
本文采用4-甲基苯酐为原料,通过氨化反应、脱水反应、NBS反应、磷盐反应、Wittig反应合成了4-(3,4,5-三氟苯撑乙烯基)邻苯二甲腈;以3,6-二羟基邻苯二甲腈、4-硝基邻苯二甲腈为原料,碱性催化剂无水碳酸钾的催化作用下,在DMF(N,N-二甲基甲酰胺)溶剂中经过亲核取代反应,分别合成了中间配体3,6-二辛氧基邻苯二甲腈以及胆固醇邻苯二甲腈;然后,通过DBU液相碱催化法合成了3种目标金属酞菁衍生物。
对一系列中间产物和目标产物的结构进行了红外、核磁等表征,并通过紫外、荧光、偏光显微镜、X-射线衍射分析(XRD)等对其进行了光物理性能与聚集态的研究。
结果表明:在紫外-可见光吸收光谱中,3种取代金属酞菁Q带出现在了不同波长位置:烷氧基酞菁铜Q带吸收出现在760nm左右;而胆固醇酞菁锌、氟代酞菁锌分别出现在了680nm、690nm左右,相对于烷氧基酞菁铜发生了蓝移。
荧光发射光谱中,烷氧基酞菁铜的最大发射波长在860nm附近,胆固醇酞菁锌最大发射波长在720nm附近,氟代酞菁锌最大发射波长在710nm附近。
并且随着金属酞菁溶液浓度的降低,荧光光谱最大发射波长发生了蓝移。
在紫外吸收光谱中,金属酞菁在不同浓度同一溶剂中,Q带吸收强度发生了改变,但没有产生明显的红移或蓝移;而同一浓度不同溶剂中,相对于极性弱的溶剂,Q带发生了蓝移。
在此基础上,同时分析了金属酞菁在溶剂中的聚集态形式,在稀溶液中以单分子和H聚集为主。
使用偏光显微镜观察金属酞菁的聚集态。
在自然光下,发现烷氧基酞菁铜聚集成针状或棒状,而胆固醇酞菁锌呈现颗粒状,转换偏光观察并未发现晶体,这与取代基有着密不可分的关系。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
金属酞菁的合成及表征
摘要:本实验是以苯酐-尿素法合成酞菁钴,以邻苯二甲酸酐、无水CoCl2、尿素为原料,以(NH4)2MoO4为催化剂,采用金属模版法合成酞菁钴,用浓硫酸再沉淀法提纯产物,纯产物通过红外光谱、紫外可见光谱进行表征。
关键词:苯酐-尿素;酞菁钴;合成;光谱测定
1 引言
酞菁类化合物是四氮大环配体的重要种类,酞菁是一个大环化合物,环内有一个空穴,可以容纳铁、钴、铜等金属元素,并结合生成金属配合物。
金属原子取代了位于该平面分子中心的两个氢原子。
由于与金属元素生成配位化合物,所以在金属酞菁分子中只有16个π电子,又由于分子的共轭作用,与金属原子相连的共价键和配位键在本质上是等同的。
故酞菁类化合物具有高度共轭π体系。
它能与金属离子形成金属酞菁配合物,其分子结构式如图。
这类配合物具有半导体、光电导、光化学反应活性、荧光、光记忆等特性。
金属酞菁是近年来广泛研究的经典金属类大环配合物中的一类,其基本结构和天然金属卟啉相似,具有良好的热稳定性,因此金属酞菁在光电转换、催化活性小分子、信息存储、生物模拟及工业染料等方面有重要的应用。
金属酞菁的合成方法主要是模版法,即通过简单配体单元与中心金属离子的配位作用,然后再结合成金属大环配合物,金属离子起模版作用。
金属酞菁的分子结构
合成反应途径如下(以邻苯二甲酸酐为原料):
2 实验内容与步骤
2.1仪器与试剂
仪器:台秤、研钵、三颈瓶(250ml)、空气冷凝管、圆底烧瓶(100mL)、铁架台、玻璃棒、抽滤瓶、布氏漏斗、可控温电热套(250mL)、电炉、温度计、抽滤瓶 DZF-III型真空干燥箱 SHZ-III型循环水真空泵、紫外─可见分光光度计
试剂:邻苯二甲酸酐、尿素、钼酸铵、无水CoCl
煤油、无水乙醇、2%盐
2、
酸、氢氧化钠溶液、蒸馏水
2.2 酞菁钴粗产品的制备
称取邻苯二甲酸酐3.69g,尿素5.95g和钼酸铵0.25g于研钵中研细后加入0.85g无水氯化钴,混匀后马上移入250ml三颈瓶中,加入60ml煤油,加热(200℃)回流2h左右,在溶液由蓝色变为紫红色后停止加热,冷却至70℃左右,加入10到15ml无水乙醇稀释后趁热抽滤。
并用乙醇洗涤2次,丙酮洗涤1次,得粗产品。
2.3 粗产品提纯
将滤饼加入2%盐酸加热煮沸后趁热抽滤,再将滤饼加入去离子水,煮沸后趁热抽滤,滤饼再加入适量氢氧化钠碱液煮沸抽滤,重复上述步骤2次,直至滤液接近无色。
将产品放在表面皿上在70℃真空干燥8h。
2.4 样品的表征与分析
干燥好后取少量样品溶于二甲基亚砜中,做紫外可见光谱分析。
3 结果和讨论
3.1 数据处理
原料:邻苯二甲酸酐3.69g ,尿素5.95g ,钼酸铵0.25g ,无水氯化钴0.85g 产品: 酞菁钴1.58g 产率:44.38% (以邻苯二甲酸酐计算产率)
3.2讨论
(1)在回流过程中空气冷凝管和三颈瓶的上部出现了白色的结晶,但是溶液却一直沸腾,白色结晶应该为尿素晶体,溶液沸腾但是三颈瓶上部还是出现结晶主要原因是加热温度不够高,蒸汽量少,没有进入空气冷凝管前一部蒸汽就因为和三颈瓶壁接触冷凝出现尿素晶体;如果加热温度调节的太高,在空气冷硬管上部就会有白色烟雾冒出,应是加热温度太高,使尿素挥发的缘故。
(2)在最后一次抽滤完成的滤饼的表面有一些白色的滤饼纤维覆盖在产品表面,应是在粗产品提纯的过程中,洗涤滤饼上产品的时候,刮下来的滤纸纤维,且每次洗涤滤饼的时候,总会有部分产品粘附在滤纸上无法完全洗涤下来,造成了产品的产率降低。
(3)在第一次加入无水乙醇稀释,趁热过滤后用丙酮洗涤时,滤液表面浮游很多油状小液滴,应是反应中用到的溶剂煤油的缘故,煤油是有机物,溶于乙醇和丙酮等有机溶剂。
4 数据附录
酞菁钴的紫外可见光谱 200400600800
2
4
6
B
A (nm)
从图可见,在600~700nm 范围内有两个酞菁类化合物所具有的特征吸收带。
金属
酞菁配合物在紫外可见光区有两个特征吸收带Q 带和B 带,Q 带在可见光区600-800nm ,B 带在近紫外区300-400nm.
酞菁钴的红外可见光谱: 40003000200010000
50
100
150
200
B
T (%) (cm -1
)
分析:
1、 金属酞菁特征吸收带主要分布在四个区域,在3030cm-1处的一组峰是芳环上
的C-H ,芳环上的C-H 的强度一般较脂肪族的C-H 弱,但谱带较尖锐.
2、 在1585 cm-1 和1610cm-1都各有一个吸收峰,这是由于芳香环上C=C 以及C=N
的伸缩振动引起的.
3、 在远红外去,骨架振动吸收带主要出现在150cm-1至200cm-1之间,对于Fe 、
Co 、Ni 、和Cu 金属酞菁,这组谱带为金属-配体-配体振动.
参考文献:
[1] 南开大学化学系中级无机化学实验编写组.中级无机化学实验,天津:南开大学出版社,1995
[2] 陈迪生.酞菁铜合成工艺的改进。
安徽化工,1996
[3] 北京大学化学系物理化学教研室;物理化学实验。
[4] 唐恢同.有机化学的光谱奠定.北京:北京化学出版社,2000。