CREBBP基因突变所致Rubinstein-Taybi综合征1例
中国人群非综合征型唇腭裂遗传背景研究进展

摘要:非综合征型唇腭裂(NSCL/ P)是一种复杂的多基因遗传性疾病,其发病机制与遗传及环境因素有关。
NSCL/ P 有关的遗传背景主要有易感基因、单核苷酸多态性。与中国人群NSCL/ P 的易感基因有关的主要有ATP
结合转运4 基因、肌腱膜纤维肉瘤癌基因同源物B、RhoGTP 酶激活蛋白29、颗粒感头状基因3、甲状腺腺瘤相关基
现FOXE1 基因的 、 rs3758249 rs10217225 和 显的异质性。新发现的区域包括:2p25. ,1 4p16. 2,
位点与 发生相关。 等 的 , , , , , , rs4460498
NSCL / P
Liu [22]
4q28. 1 5p12 6p24. 3 8p11. 23 8q22. 1 9q22. 32
能与样本的数量及种群的差异性有关。
RubinsteinTaybi 综合症的发生有关,这是一种常染
1. 5 THADA THADA 位于染色体2p21 区域,有 色体显性遗传病,面部特征表现伴有唇裂或者腭裂。
38 个外显子,长度370 。kb THADA 最早被发现是 、 、 和 等染色体区域 10q25. 3 17p13. 1 20q12 1q32. 2
内对NSCL/ P 的研究多着眼于IRF6、MTHFR、 为ABCA4 基因的突变常导致了各类遗传性视网膜
、 、 、 、 等与 MSX1 TGFα RARα BCL3 TGFβ3
NSCL / P
关系密切的传统“热门片段”,研究手段通常采用限
变性疾病的发生[4]。 Huang 等[5] 研究认为, rs560426 不是我国南方汉族人群NSCL/ P 的易感位
甲状腺腺瘤的靶基因,与甲状腺腺良性肿瘤有 与中国人NSCL/ P 发病有关。染色体10q25. 3 中的
【2017年整理】克雅氏病一例

Creutzfeld-Jakob病 1例陈龙华李宏军卢禹病例资料患者,女,59岁,农民,以进行性痴呆,言语不利,行走不稳4个月为主诉入院。
4 个月前记忆力减退,注意力下降,伴随行走不稳。
3 个月前认知障碍明显,不认识亲属,并伴随言语困难,四肢僵硬,大小便失禁。
血压、血脂、血糖正常。
无恶心、呕吐、发热。
查体:神清,淡漠,计算力、记忆力差,构音障碍,语言表达困难,双瞳孔等大等圆,直径约3mm ,对光反射灵敏,口角无歪斜,伸舌居中,四肢肌力、肌张力、腱反射均无异常,指鼻试验及跟膝胫试验稍欠稳准,脑膜刺激征阴性。
头颅CT见双侧尾状核头部、体部及壳核对称性略低密度改变,边缘模糊(图1)。
头颅MRI示:双侧尾状核、壳核对称长T2信号(图2),FLARI像及DWI序列均为高信号(图4、5),T1加权像呈略低信号(图3),增强扫描无强化(图6)。
脑脊液检测:14-3-3 蛋白(Western blot)阳性。
PRNP基因序列分析:1.E200K 突变;2.129位氨基酸多态性为M/M型。
动态脑电图:3相棘-慢复合波。
诊断为:遗传型Creutzfeldt—Jakob 病。
讨论海绵样变脑病(Creutzfeldt—Jakob disease,CJD),是一种人畜共患、中枢神经系统慢性非炎症性致死性疾病,罕见。
1920年由德国神经病学家Creutzfeldt 和 Jakob首次报道。
目前关于CJD 的发病有两种假说:外源性感染和正常PrP 基因的点突变[1]。
即携带朊蛋白的动物和少数医源性感染和遗传的朊蛋白基因突变所致。
CJD的病理学改变局限在中枢神经系统,包括海绵样变性、星形胶质细胞增生和神经元丢失;组织病理学诊断特征是灰质海绵样变性,以神经元和胶质细胞的单个或聚集分布的空泡为标志;病变弥漫于大脑灰质、纹状体、丘脑、脑干的灰质结构和小脑皮质的分子层,也可分布在海马[2]。
随着病情进展,脑萎缩变明显。
本病发病高峰为50~70岁,临床表现多样性,以人格改变起病,伴进行性智力衰退,无发热,可以出现不同程度的神经系统症状,如肌阵挛和共济失调、癫痫等,晚期均发展为去皮层状态。
Rubinstein-Taybi综合征一例报告

6·罕少疾病杂志 2021年12月 第28卷 第 6 期 总第149期【第一作者】耿 健,男,主治医师,主要研究方向:儿童重症早期康复干预。
E-mail:********************【通讯作者】孙爱梅,女,主任医师,主要研究方向:儿童脑损伤疾病的超早期诊治。
E-mail:****************·罕见病研究·短篇 R u b i n s t e i n -Ta y b i 综合征(R u b i n s t e i n -Ta y b i syndrome,RSTS)是一种罕见的常染色体显性遗传病,其在人群中的发病率为1∶100000~1∶125000[1],在智力发育迟缓患者中为1∶500[2]。
患儿的主要临床表现为特殊面容、发育迟缓、智力低下及宽大拇指、足趾等。
由于RSTS的临床表现存在异质性,且尚未形成全面而统一的临床标准,大部分的RSTS病例都需要借助基因检测以明确诊断[3]。
截至目前,已经明确约50%~70%由CREBBP基因突变引起,为RSTS-1,10%由EP300基因突变所致,为RSTS-2,20%~40%尚未明确致病基因[4]。
本研究通过对1例疑似Rubinstein-Taybi综合征患儿的临床表现进行分析,并运用全外显子捕获测序进行基因检测,同时参考HGMD pro及ACMG分级,最终在CREBBP基因外显子区发现一处杂合突变点:c.4268dupC,且证实其为该病的发病原因,用Sanger测序法验证患儿和父母。
分析患儿的基因型,并评估康复治疗效果。
1 病历资料 患儿,女,3岁10月,以“至今3岁10月认知差,发音少”为代主诉入院。
患儿系G3P3,孕7月无明显诱因见红,未治疗,因胎膜早破于孕38+4周剖宫产娩出。
生后哭声弱,Apgar评分不详,出生体重3.4kg。
生后即发现其小头,头发、眉毛浓密而黑,发际线低。
自幼生长发育缓慢,5月竖头稳,9月会叫“妈妈”,10月会坐,1岁8月会走。
PHIP基因突变所致Chung-Jansen综合征一例
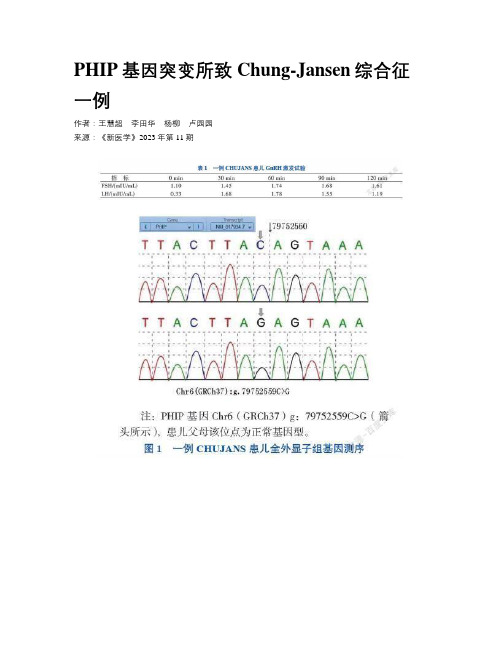
PHIP基因突变所致Chung-Jansen综合征一例作者:王慧超李田华杨柳卢园园来源:《新医学》2023年第11期【摘要】 Chung-Jansen综合征(CHUJANS)是一种常染色体显性遗传病,是新近发现的罕见肥胖综合征,主要表现为发育迟缓、智力障碍、肥胖和畸形。
该文报道1例以肥胖、睾丸小为主要表现的CHUJANS患儿,该患儿发育迟缓、智力障碍,伴有左肾缺如及低促性腺激素性性功能减退,基因检测结果提示PHIP基因突变,突变位点c.600+1G>C,最终诊断为CHUJANS。
经过长期综合性治疗,患儿远期生活质量获得极大改善。
CHUJANS发病率低,且累及多系统,该例扩展了CHUJANS的基因突变谱,有助于提高临床医师对该疾病的认识水平,及早识别并给予干预将有助于改善患者预后。
【关键词】 Chung-Jansen综合征;PHIP基因;杂合突变;儿童A case of Chung-Jansen syndrome caused by PHIP gene mutation Wang Huichao△, Li Tianhua, Yang Liu, Lu Yuanyuan. △970 Hospital of the PLA Joint Logistic Support Force,Weihai 264299, ChinaCorresponding author, Lu Yuanyuan, E-mail:*******************【Abstract】 Chung-Jansen syndrome (CHUJANS), an autosomal dominant genetic disorder, is a newly discovered rare obesity syndrome, mainly manifesting as developmental delay, mental retardation, obesity and dysmorphism. We reported one CHUJANS child with obesity and small testes as the main manifestations. The patient had developmental delay, mental retardation, complicated with left renal agenesis and hypogonadotropic hypogonadism. Genetic testing prompted PHIP gene mutation at c.600+1G>C. The child was diagnosed with CHUJANS. After long-term comprehensive treatment, the long-term quality of life was significantly improved. As Chung-Jansen syndrome is low in prevalence and multi-systemic, this case report expands the spectrum of mutations in CHUJANS,which can deepen clinicians’ understanding of this disease. Early diagnosis and intervention contribute to enhancing clinical prognosis.【Key words】 Chung-Jansen syndrome; PHIP gene; Heterozygous mutation; ChildrenChung-Jansen综合征(CHUJANS,OMIM#617991)是一种以发育迟缓、智力障碍、肥胖和畸形为特征的常染色体显性遗传病,由位于6q14染色体上的PHIP基因(OMIM#612870)中的杂合突变所致,可在婴儿期发病,大多为基因突变从头合成,少数为家族遗传[1]。
1例Krabbe病患儿的临床资料及家系基因变异分析

1例Krabbe病患儿的临床资料及家系基因变异分析张坡;陈秋;姜跃彭;童鸣;胡苏玮【期刊名称】《临床检验杂志》【年(卷),期】2024(42)5【摘要】球形细胞脑白质营养不良(globoid cell leukodystrophy,GLD,OMIM 245200)又称Krabbe病,是一种罕见的溶酶体贮积病,为常染色体隐性遗传病。
该病是由于半乳糖脑苷脂酶(galactocerebrosidase,GALC)基因缺陷,导致溶酶体内半乳糖脑苷脂酶活性缺失,引起半乳糖脑苷脂不能降解而蓄积,产生鞘氨醇半乳糖苷毒性,造成中枢及外周神经纤维髓鞘脱失、星形胶质细胞增生及巨噬细胞浸润,从而引起一系列中枢与周围神经系统症状[1]。
Krabbe病根据发病年龄分为4种临床类型,其中婴儿型在出生后6个月内发病,90%病例为婴儿型,患儿大多在2岁左右夭折;晚发婴儿型的发病时间在19个月至4岁之间;少年型在4岁至19岁之间发病;成人发病在20岁后。
Krabbe病在国内罕见,多为个例报道,欧美国家发病率约为1/100000[2]。
现将1例婴儿型Krabbe病患儿的临床表现和家系基因变异分析报告如下。
【总页数】3页(P389-391)【作者】张坡;陈秋;姜跃彭;童鸣;胡苏玮【作者单位】扬州大学医学院附属扬州妇幼保健院医学遗传中心【正文语种】中文【中图分类】R722.11【相关文献】1.青少年型Krabbe病的临床、影像学特点及酶学检测(附1家系报告)2.遗传性压力易感性周围神经病一家系临床资料及基因分析3.PIK3CD基因变异致活化的PI3Kδ综合征一例临床资料及基因变异分析4.表现为单纯痉挛性截瘫的青少年型Krabbe病一例遗传及临床分析5.巨轴索神经病临床、神经病理和GAN基因变异特点(附1家系报告)因版权原因,仅展示原文概要,查看原文内容请购买。
班替综合征1例报告并文献复习

班替综合征1例报告并文献复习班替综合征(Bannayan-Riley-Ruvalcaba综合征,BRRS)是一种罕见的遗传性疾病,也称肿瘤性常染色体显性隐性遗传综合征,多见于婴儿至儿童期,至今尚无特效治疗。
本文报道了一例BRRS患儿的一般资料、临床表现和治疗情况。
一、病史本病例患儿男性,现年6岁,来自广西壮族自治区的一家三口。
患儿生于2013年,在出生时发现头部与面部畸形,结合患儿的性别、家族史和身体特征,认为可能患有BRRS。
二、临床表现患儿出生后,发现头部与面部畸形,伴有大耳、肿瘤性皮疣、黄疸、染色体异常及其他神经系统症状。
体格检查时发现,头围增大,眼眶有轻度膨隆,耳朵大、头先出,眉毛稀疏;上颌骨发育不良,口腔黏膜及牙齿未见明显异常;体重及身高偏低,发育迟缓;脐带周围褶皱;皮肤下出现多发性肿瘤性皮疣,大小不一;腹部增大,腹壁肌肉偏薄;足跟缩短。
无家族遗传史,患儿母亲经常产抑郁症。
三、诊断根据上述特征,本病例诊断为BRRS,患儿经过基因检测结果证实,携带PTEN基因突变,诊断结果符合BRRS的诊断标准。
四、治疗本病例患儿,采用理疗加营养补充治疗,遵医嘱给予复方熊果素、复方小儿多维元素、复方小儿益生菌等营养支持,定期复查,观察病情变化。
五、结论BRRS是一种罕见的常染色体显性遗传综合征,其临床表现比较复杂,具有不可逆转的潜在危险性。
本病例经基因检测后证实患儿携带PTEN基因突变,诊断结果符合BRRS的诊断标准,治疗上采取理疗加营养补充治疗,定期复查,观察病情变化。
本病例服用熊果素、小儿多维元素、小儿益生菌等营养支持的治疗效果良好,病情有所缓解,表明上述治疗方法对治疗BRRS有一定的辅助治疗作用,可为临床管理提供一定的参考价值。
然而,因BRRS病理机制较为复杂,治疗目前尚无特效治疗,生物学研究也仍存在不足之处,因此,仍需要进一步深入探究。
在家庭环境的影响下,也需要积极的家庭教育及心理支持,积极采取拉线管理,加强定期复查,防治病情发展,以全面促进患儿的正常发育与成长。
【案例分享】Gilbert综合征
【案例分享】Gilbert综合征疾病概述Gilbert综合征(Gilbert Syndrome,GS)⼜称为体质性肝功能不良性黄疸,是由于肝组织摄取⾮结合胆红素障碍或微粒体内葡萄糖醛酸转移酶不⾜,致使⾎液中⾮结合胆红素显著增⾼⽽发⽣黄疸的⼀类较常见的遗传性代谢疾病,1901年Gilbert⾸先报告。
Gilbert综合征临床表现特点为长期间歇性轻度黄疸,多⽆明显症状,在临床中易被误诊为肝炎。
发病率⼤约为5%左右,男性多见,男⼥发病⽐例1.5 :1到7 :1,以青年期(15~20岁)发病最多见,可因紧张、劳累、饮酒、感染、受凉、腹泻、便秘、饥饿、或合并其他疾病⽽加重或诱发。
⼀般情况良好,黄疸加重可有乏⼒、消化不良、肝区不适等症状。
⽪肤和巩膜轻中度黄染是唯⼀的体征。
⾎胆红素波动在1~3mg/dl,⾼或低于此值也常看到。
轻型⼀般不超过5mg/dl,重型可超过5mg/dl。
发病机制在肝脏中⾮结合胆红素的葡萄糖醛酸化是胆红素转化、分泌、排泄的重要条件,GS患者有胆红素的⽣成、摄取、转运和结合的障碍,主要表现为尿苷⼆磷酸葡萄糖醛酸转移酶(UDP2glucuronosyltransferase,UGT)活⼒明显下降,仅及正常⼈的30%。
UGT主要集中在肝内质⽹和核膜。
UGT的活性依赖于⼤量同⼯酶的存在,它以尿苷-5⼆磷酸葡萄糖醛酸(UGA)为糖基供应体与底物结合,增加内源性和外源性物质的亲⽔性,使其更易于随尿与胆汁排出体外。
该酶催化UDP2葡萄糖醛酸酶分⼦中的葡萄糖醛酸向游离胆红素的丙酸基转移,游离胆红素进⼊肝细胞后,被肝细胞浆内的两种低分⼦可溶性“受体蛋⽩”(Y,Z蛋⽩接受)带到滑⾯内质⽹,在酶的作⽤下进⾏结合,形成胆红素单葡萄糖醛酸和胆红素双葡萄糖醛酸。
根据其cDNA序列的同源性将UGT超基因家族分为四个亚族:UGTl、UGT2、UGT4、UGT8。
⼈类UGTl基因定位于染⾊体2q37,包括5个外显⼦,第l外显⼦区域5’端由13个可替换的第1外显⼦(UGTlA)组成。
遗传性球形红细胞增多症合并Gilbert综合征1例报告
遗传性球形红细胞增多症合并Gilbert综合征1例报告张玉姣;郑英;王秀红;蔡颖;马安林
【期刊名称】《临床肝胆病杂志》
【年(卷),期】2022(38)4
【摘要】1病例资料患者男性,26岁,因“反复全身皮肤及巩膜黄染26年”于2021年6月24日收住中日友好医院。
患者在出生5个月时出现全身皮肤、巩膜黄染,尿色加深,未予重视。
12岁时,再次因“皮肤及巩膜黄染”就诊于解放军总医院,腹部超声提示:慢性胆囊炎、胆囊结石(充满型)、脾肿大,未明确诊断。
后就诊于北京儿童医院,查肝功能:DBil 38μmol/L,IBil 206.3μmol/L,ALT、AST、GGT、ALP均正常,血常规.
【总页数】3页(P888-890)
【作者】张玉姣;郑英;王秀红;蔡颖;马安林
【作者单位】中日友好医院感染疾病科;中日友好医院病理科;中日友好医院检验科;解放军联勤保障部队第九四〇医院消化内科
【正文语种】中文
【中图分类】R65
【相关文献】
1.遗传性球形红细胞增多症合并G6PD缺乏1例报告
2.遗传性球形红细胞增多症误诊为Gilbert综合征1例
3.非活动性HBsAg携带者合并遗传性球形红细胞增多
症1例并文献复习4.Gilbert综合征合并遗传性球形红细胞增多症1例5.终末期肝病合并遗传性球形红细胞增多症并存地中海贫血1例
因版权原因,仅展示原文概要,查看原文内容请购买。
Rubinstein-Taybi综合征3例临床及遗传学分析
第59卷 第1期2023年02月青岛大学学报(医学版)J O U R N A LO FQ I N G D A O U N I V E R S I T Y (M E D I C A LS C I E N C E S)V o l .59,N o .1F e b r u a r y2023[收稿日期]2021-05-19; [修订日期]2022-10-30[基金项目]山东省医药卫生科技发展计划项目(2018W S 442)[第一作者]杨宁(1980-),女,硕士,副主任医师,硕士生导师㊂E -m a i l :1183054587@q q.c o m ㊂R u b i n s t e i n -T a yb i 综合征3例临床及遗传学分析杨宁,顾晓冉,白丽君(德州市人民医院儿科,山东德州 253000)[摘要] 目的 探讨R u b i n s t e i n -T a yb i 综合征(R S T S )的临床表型及遗传学特点㊂方法 回顾性分析2019年我院门诊确诊的3例R S T S 病儿的临床资料㊂结果 3例病儿均有特殊面容㊁拇指和大趾宽扁㊁体格及智力发育迟滞等特征㊂分子遗传学检测显示,3例致病基因均为C R E B B P ,其中1例携带C R E B B P 基因c .4890+2T>C 杂合变异,为新突变;2例为16p 13.3连续缺失,为已知致病突变㊂结论 对于存在特殊面容㊁宽拇指/趾和生长发育迟缓的病儿应考虑R S T S 的可能,C R E B B P 为该病的主要致病基因之一,基因靶向检测和基因组检测有助于R S T S临床确诊㊂[关键词] R u b i n s t e i n -T a y b i 综合征;疾病特征;基因检测[中图分类号] R 725.96 [文献标志码] B [文章编号] 2096-5532(2023)01-0139-04d o i :10.11712/jm s .2096-5532.2023.59.032[开放科学(资源服务)标识码(O S I D )][网络出版] h t t ps ://k n s .c n k i .n e t /k c m s /d e t a i l /37.1517.R.20230308.1057.015.h t m l ;2023-03-09 16:56:31C L I N I C A LA N DG E N E T I CF E A T U R E SO FR U B I N S T E I N -T A Y B I S Y N D R O M E :A NA N A L Y S I SO FT H R E EC A S E S Y A N GN i n g ,G U X i a o r a n ,B A IL i ju n (D e p a r t m e n t o f P e d i a t r i c s ,D e z h o uP e o p l e sH o s i p i t a l ,D e z h o u253000,C h i n a )[A B S T R A C T ] O b je c t i v e T o i n v e s t i g a t e t h e c l i n i c a l p h e n o t y p e a n d g e n e t i cf e a t u r e s o fR u b i n s t e i n -T a y b i s y n d r o m e (R S T S ).M e t h o d s Ar e t r o s p e c t i v e a n a l y s i sw a s p e r f o r m e d f o r t h e c l i n i c a l d a t a o f t h r e e c h i l d r e nw h ow e r e d i a gn o s e dw i t hR S T S i n t h e o u t -p a t i e n t s e r v i c e o f o u r h o s p i t a l i n 2019. R e s u l t s A l l t h r e e c h i l d r e n h a d t h e f e a t u r e s o f u n u s u a l f a c i e s ,b r o a d t h u m b s a n d b i gt o e s ,a n d p h y s i c a l a n dm e n t a l r e t a r d a t i o n .T h em o l e c u l a r g e n e t i c a n a l y s i s s h o w e d t h a t t h e C R E B B P g e n ew a s t h e p a t h o ge n i c g e n e i n a l l t h r e e c h i l d r e n ,a m o n g w h o mo n e c h i l d c a r r i e d t h e c .4890+2T>Ch e t e r o z y g o u sm u t a t i o n of t h e C R E B B Pg e n e ,whi c hw a s a n o v e l m u t a t i o n ,a n d t w o c h i l d r e nh a d16p 13.3c o n t i n u o u s d e l e t i o n ,w h i c hw a s ak n o w n p a t h o ge n i cm u t a t i o n . C o n c l u s i o n T h e p o s s i -b i l i t y o fR S T Ss h o u l db e c o n s i d e r e df o r c h i l d r e nw i t hu n u s u a l f a c i e s ,w i d e t h u m b s /t o e s ,a n dg r o w th r e t a r d a ti o n .C R E B B P i s o n e o f t h em a i n p a t h o g e n i c g e n e s f o r t h i s d i s e a s e ,a n d t a r g e t e d g e n e d e t e c t i o n a n d g e n o m e d e t e c t i o nm a y h e l p w i t h t h e c o n f i r m e d d i a g-n o s i s o fR S T S i n c l i n i c a l pr a c t i c e .[K E Y W O R D S ] R u b i n s t e i n -T a y b i s y n d r o m e ;d i s e a s e a t t r i b u t e s ;g e n e t i c t e s t i n gR u b i n s t e i n -T a y b i 综合征(R S T S )是一种极为罕见的遗传综合征,为多发性先天性异常,发病率为1/100000~1/125000,发病无性别差异,目前还没有明确的诊断标准,其特征性的临床表现为智力低下㊁出生后生长发育落后㊁特殊面容㊁宽拇指/趾等㊂1991年首次确定了该病的遗传基础,目前明确的致病基因为C R E B B P 和E P 300,由C R E B B P 基因突变所致者为1型(R S T S 1),占该综合征的50%~70%,由E P 300基因突变所致者为2型(R S T S 2),占该综合征的5%~8%,但仍有约30%未找到致病基因[1-2]㊂2019年我院门诊通过分子遗传学检测方法确诊R S T S 病儿3例,本研究对其临床及遗传学特征进行分析,并复习相关文献,探讨R S T S 临床表型与基因型的关系㊂1 病例报告例1,男,7岁7个月㊂因发育迟缓就诊㊂病儿系第1胎第1产,足月顺产出生,出生体质量3.2k g ,身长50c m ,出生时无窒息,出生后身高增长较慢㊂病儿生后3个半月不能竖头;8个月给予头颅M R I 检查示脑白质发育不良;3岁开始说话,不听指令,认知理解力差,构音不清㊂父母非近亲结婚,身体均健康,否认类似疾病家族史㊂体格检查示:身高115c m ,体质量23.7k g ,头围51.5c m ,身材矮小,鬼脸笑容,弓形眉,睫毛长,鼻梁宽阔,鼻中隔呈喙状,高腭弓,隐睾,四肢肢端肥大,双手通贯掌,心肺腹查体无异常,四肢肌力㊁肌张力未见异常(图1A~C )㊂实验室检查:血尿常规㊁促甲状腺激素及促肾上腺皮质激素㊁遗传代谢病筛查等均未见异常㊂脑电图检查示基本节律慢化㊂韦氏儿童智力量表检测:语言智商41,操作智商<40,全量表智商<40㊂全外显子组测序C N V 检测(W E S -C N V )显示,16号染色体16p 13.3处缺失0.60M b 大小区段,位置为c h r 16:3458315-4057564,此区段覆盖了导致R S T S 的全部基因,包含单倍剂量敏感基因C R E B B P 以及S L X 4和D N A S E 1基因㊂例2,女,3岁8个月㊂因发育迟缓就诊㊂病儿系第2胎第2产,足月剖宫产,出生时无窒息,体质量3k g ,生后存在动脉导管未闭,后自行关闭,3岁2个月时因泪道阻塞行手术Copyright ©博看网. All Rights Reserved.140青 岛 大 学 学 报 (医 学 版)59卷治疗;现不会说话,不会坐,不会独走㊂体格检查示:身高95c m ,体质量14.1k g ,头围43.5c m ,头围小,弓形眉,双侧上睑下垂,鼻梁宽阔,鼻中隔呈喙状,耳位低,小下颌,高腭弓,四肢肢端肥大(图1D ㊁E )㊂G e s e l l 测评:适应性22,大运动28,精细动作22,语言13,个人-社交16㊂a C G H+S N P 芯片检测示3处染色体异常:①14号染色体q 32.33扩增,大小约550k b ,位置为c h r 14:(106313878-106863507)ˑ3,在正常人染色体拷贝数变异多态数据库中有收录,暂未见病理性报道;②16号染色体p 13.3扩增,大小约3.642M b,位置为c h r 16:(858800-3728099)ˑ3,该扩增的临床表现为骨骼畸形和发育迟缓;③16号染色体p 13.3缺失,大小约981k b ,位置为c h r 16:3796442-4776992,包含C R E B B P ㊁A D C Y 9和S R L 基因,该缺失与R S T S 有关㊂例3,男,1岁8个月㊂因发育迟缓就诊㊂病儿系第1胎第1产,双绒双羊,双胎之大,36+1周剖宫产,体质量2.4k g ,出生时无窒息,生后8个月会坐,1岁时因肺炎链球菌化脓性脑膜炎住院治疗;现不会说话,坐不稳,不会爬,不会独走㊂体格检查:身高86.3c m ,体质量12.83k g ,头围47c m ,弓形眉,耳位低,鼻梁宽阔,鼻中隔呈喙状,隐睾,四肢肢端肥大(图1F ~H )㊂13个月时头颅M R I 示双侧额颞叶脑萎缩,胼胝体薄,胼胝体嘴形态欠规则,幕上脑室扩张,DW I 可见双侧额颞蛛网膜下隙内条片状等高信号㊂G e s e l l 测评:适应性61,大运动71,精细动作49,语言34,个人-社交40㊂疾病外显子组-订制捕获测序结果示位于16号染色体p 13.3的C R E B B P 基因c .4890+2T>C 变异㊂经检测病儿父母均不携带该变异㊂双胎之小发育正常㊂A :例1颅面特征;B :例1鬼脸笑容;C :例1手指肢端肥大;D :例2颅面特征;E :例2手指肢端肥大;F :例3颅面特征;G ㊁H :例3拇指和脚趾粗大㊂图1 3例病儿的颅面特征和肢端肥大表现2 讨 论R S T S 是一种常染色体显性遗传病,1963年由R U B I N -S T E I N 等[3]首次报道,该病以智力低下㊁拇指和脚趾粗大以及面部异常为特征㊂直到90年代,该综合征诊断仍然只限于临床和放射学(手和脚的X 线片)诊断㊂1991年,P E T R I 等首次通过报告R S T S 病人染色体16p13.3区域出现断点的平衡易位,证明了R S T S 的遗传起源㊂C R E B B P (16p 13,OM I M#600140)和E P 300(22q13,OM I M#402700)这两个普遍表达且高度同源基因的改变是R S T S 的遗传基础,分别导致R S T S 1和R S T S 2[4]㊂60%的R S T S 病例C R E B B P 基因发生新的变异,C R E B B P 是与该综合征相关的最重要的基因[5]㊂到目前为止,在C R E B B P 中已有413个独特的致病变异体被描述,而在E P 300中只有127个㊂R S T S 的临床表现多样,目前没有明确的诊断标准,但该病有一系列特征性的临床表现,如独特的面部特征(包括眉毛高拱形,睫毛长,眼睑裂下垂,鼻梁宽阔,鼻中隔呈喙状,上颚高拱形,轻度小颌畸形,有典型的鬼脸或不正常的微笑)㊁中度至重度智力障碍㊁身材矮小㊁拇指和拇趾粗大且通常有角度[2]㊂产前生长通常是正常的,出生时的生长参数正常或接近正常,然后身高㊁体质量㊁头围百分位数在生命的头几个月迅速下降,身材矮小是成年病人的典型特征,肥胖可Copyright ©博看网. All Rights Reserved.1期杨宁,等.R u b i n s t e i n-T a y b i综合征3例临床及遗传学分析141能发生于儿童或青少年时期[6]㊂平均智商为35~50,然而,发育结果差异很大,一些患有E P300-R S T S的病人智力正常[7]㊂尿路结构异常很常见,几乎所有男性都有隐睾[2]㊂约1/3的病人患有各种类型的先天性心脏病,其他特征包括眼部异常㊁听力丧失㊁呼吸困难㊁进食问题㊁反复感染㊁严重便秘和肿瘤发生风险增加[2]㊂目前尚没有发现可靠的基因型-表型相关性[8]㊂然而,在R S T S病人中,低智商㊁自闭症可能与大量基因缺失相关联[9]㊂R S T S2似乎比R S T S1具有更温和的表型㊂与R S T S1病人相比,R S T S2病人面部畸形程度较轻,认知功能较好,但可能有更严重的小头畸形和面部骨骼结构畸形[10]㊂超过99%的R S T S1病人都存在C R E B B P基因的新杂合子突变或缺失[10]㊂R S T S1有3种不同的表型,即经典型㊁轻度型和非常严重型[11]㊂经典型通常是由C R E B B P基因的缺失或截断突变引起的,具有典型的临床表现㊂轻度型(也称为不完全R S T S1)通常是由错义C R E B B P突变引起的,其特征是有轻微的面部特征㊁粗大拇指和拇趾,无智力障碍,其他系统性特征通常缺失[12]㊂染色体16p13.3连续缺失综合征是一种非常严重且致命的表型,除典型临床表现外,该综合征病人还表现为严重的危及生命的感染㊁严重的智力障碍㊁严重的新生儿惊厥㊁多灶性心律失常㊁左心发育不良和多脾㊂这种非常严重的表型是由基因大量缺失引起的,包括C R E B B P基因和3'邻近基因(即D N A S E1和T R A P1),包括C R E B B P基因但没有这两个3'邻近基因的大量缺失病人则表现出典型而非严重的表型[13]㊂C R E B B P基因大小约150k b,包含31个外显子,编码2442个氨基酸的C R E B结合蛋白(C B P);E P300基因也包含31个外显子,编码p300蛋白㊂这两种蛋白都作为转录辅活化因子介导R N A聚合酶Ⅱ复合物和D N A结合转录因子之间的相互作用,参与许多基本的细胞代谢过程,如D N A 修复㊁生长㊁分化㊁细胞凋亡㊁肿瘤抑制等[14]㊂此外,C B P和p300还起着表观遗传因子作用,通过介导组蛋白乙酰转移酶(HA T)活性改变染色质结构并调节基因表达,HA T活性的丧失与R S T S之间存在直接联系[15-16]㊂有动物实验结果显示,纯合子C RE B B P或者E P300基因敲除小鼠在出生前死亡,双杂合子E P300+/-/C R E B B P+/-小鼠具有胚胎致死性,表明这两种基因的正确组合对胚胎发育至关重要[17-18]㊂然而,由于有400多种蛋白质相互作用,这两种基因改变导致疾病的确切机制目前尚不清楚[19-20]㊂C B P/p300作为外部刺激的检测器㊁积分器和调节器来修饰细胞表型,进一步增加了疾病的复杂性[21]㊂到目前为止,广泛的致病变异已经被报道,包括点突变㊁大量缺失以及突变中的移码㊁无义㊁错义和剪接变异,这些变异占R S T S中所发现的遗传变异的30%~50%;在约10%的R S T S病例中,大小不等的缺失(基因内缺失㊁全基因缺失或向邻近区域扩展)也是疾病原因,然后是倒置和易位;仍有25%~30%的临床怀疑为R S T S先证者的分子原因不明[14]㊂R S T S确诊主要通过分子遗传检测,包括基因靶向检测(系列单基因检测㊁多基因面板㊁染色体微阵列分析)和基因组检测(外显子组测序㊁外显子组阵列㊁基因组测序)㊂基因靶向检测要求临床医生确定可能涉及的基因,而基因组检测则不需要㊂R S T S的表型复杂多样,具有独特发现的个体很可能可以通过基因靶向检测进行诊断,而表型与许多其他遗传性矮小和(或)智力残疾疾病无法区分的个体更可能通过基因组检测来诊断,其中外显子组测序是最常用的㊂R S T S通常是由于家族中新的致病变异而导致的,大多数病例为单一个案,即家庭中唯一受影响的成员[2,10]㊂在大多数情况下,R S T S病人的父母不受影响,但由于双亲杂合或亲本体细胞和(或)种系嵌合体可能导致出现轻度表型,故推测兄弟姐妹发生R S T S的风险仍较高[22]㊂一旦在受累家庭成员中发现了致病性变体,就应该对高危妊娠进行产前检测和植入前基因诊断㊂本文报道的3例病儿,其致病基因均为C R E B B P,表型为R S T S1㊂其中例1和例2均为染色体16p13.3相邻缺失综合征,除C R E B B P基因缺失以外,例1还存在S L X4和D N A S E1基因缺失,例2还存在A D C Y9和S R L基因缺失,并且合并16p13.3基因重复,缺失与重复区域无重叠㊂例3病儿C R E B B P基因发生c.4890+2T>C变异(第4890+2号核苷酸由胸腺嘧啶变异为胞嘧啶),变异位于第29号内显子,为剪接位点突变,该位置序列在物种间高度保守,变异可导致剪接受体位点丧失,发生剪接异常,该变异在d b S N P数据库㊁千人基因组数据库㊁E x A C数据库以及g n o m A D数据库中均未见报道㊂3例病儿出生体质量㊁身长㊁头围均正常,在生后体格发育方面,例1主要存在身材矮小,例2为小头畸形,例3体质量㊁身长㊁头围均在正常范围㊂3例病儿均具有典型的面部特征及宽拇指/趾,只有例1存在鬼脸笑容,2例男孩均存在隐睾;此外,例1存在脑白质发育不良,例2存在泪道阻塞及动脉导管未闭,例3曾患肺炎链球菌脑膜炎㊂3例病儿均存在智力低下,例2尤为严重,考虑原因为基因缺失范围大,并且合并16号染色体的有害重复;3例病儿在随访过程中语言功能障碍随年龄增加更加明显㊂3例病儿均通过分子遗传学检测确诊,例1应用外显子组测序,例2应用染色体微阵列分析,例3应用多基因面板检测㊂染色体微阵列分析可筛查全基因组大范围缺失或重复,外显子组测序关注的是编码区信息,而多基因面板检测则是关注包括C R E B B P㊁E P300和其他感兴趣基因的多基因小组,可获得指定目标区域的遗传信息,与外显子组测序比较可以提高效率㊁节约成本,在实际工作中可根据疾病特征及对疾病的认识程度选择不同的分子遗传检测方法㊂目前R S T S无特效的治疗方法,由于表观遗传变化的主要特征是其可逆性,因此新的遗传和表观遗传疗法可能是治疗R S T S有希望的方法[23]㊂组蛋白去乙酰化酶(H D A C)抑制剂在R S T S治疗中具有潜在应用,针对HA T活性降低的R S T S小鼠模型的临床前研究表明,H D A C抑制剂治疗可以改善部分认知功能障碍[24]㊂有研究结果表明,超过90%的R S T S病人可以活到成年,并且大多数病人在自我照顾方面Copyright©博看网. All Rights Reserved.142青岛大学学报(医学版)59卷有一定的独立性,但超过90%的病人是需要有人监管的[2]㊂由于R S T S可能会出现许多非特异性的并发症,因此很难制定一个通用有效的随访方案,一般来说,骨科随访㊁青春期和神经精神症状期的饮食监测以及成人的眼科评估都应该是重点㊂本文3例病儿的远期生存情况㊁精神运动发育和各系统的评估等尚待进一步追踪随访㊂[参考文献][1]P E T R I FF,G I L E S R H,D A UW E R S E H G,e ta l.R u b i n-s t e i n-T a y b i s y n d r o m ec a u s e db y m u t a t i o n si nt h et r a n s c r i p-t i o n a l c o-a c t i v a t o r C B P[J].N a t u r e,1995,376(6538):348-351.[2]M I L A N ID,MA N Z O N I F M,P E Z Z A N I L,e t a l.R u b i n s t e i n-T a y b i s y n d r o m e:c l i n i c a l f e a t u r e s,g e n e t i cb a s i s,d i a g n o s i s,a n dm a n a g e m e n t[J].I t a l i a nJ o u r n a l o f P e d i a t r i c s,2015,41:4.[3]R U B I N S T E I NJH,T A Y B IH.B r o a d t h u m b s a n d t o e s a n d f a-c i a l a b n o r m a l i t i e s:a p o s s i b l em e n t a l r e t a rd a t i o n s y n d r o m e[J].A m e r i c a n J o u r n a l o fD i s e a s e so fC h i l d r e n,1963,105(6):588-608.[4]K O R Z U SE.R u b i n s t e i n-T a y b i S y n d r o m ea n dE p i g e n e t i cA l t e-r a t i o n s[J].A d v a n c e s i n E x p e r i m e n t a l M e d i c i n ea n dB i o l o g y, 2017,978:39-62.[5]S P E N A S,M I L A N I D,G E R V A S I N I C.U l t r a-r a r es y n-d r o me s:t h e e x a m p l eo fR u b i n s t e i n-t a y b i s y n d r o m e[J].J o u r-n a l o f P e d i a t r i cG e n e t i c s,2015,4(3):177-186.[6]B E E T SL,R O D RÍG U E Z-F O N S E C A C,H E N N E K AM R C.G r o w t hc h a r t s f o r i n d i v i d u a l sw i t hR u b i n s t e i n-T a y b i s y n d r o m e[J].A m e r i c a n J o u r n a l o fM e d i c a l G e n e t i c s P a r tA,2014,164A(9):2300-2309.[7]F E R G E L O TP,V A NB E L Z E N M,V A N G I L SJ,e t a l.P h e n o-t y p e a n d g e n o t y p e i n52p a t i e n t sw i t h R u b i n s t e i n-T a y b i s y n-d r o me c a u s e db y E P300m u t a t i o n s[J].A m e r i c a nJ o u r n a lo fM e d i c a lG e n e t i c sP a r tA,2016,170(12):3069-3082. [8]V A NB E L Z E N M,B A R T S C H O,L A C OM B ED,e t a l.R u b i-n s t e i n-T a y b i s y n d r o m e(C R E B B P,E P300)[J].E u r o p e a n J o u r n a l o fH u m a nG e n e t i c s,2011,19(1):3.[9]MA R A B O T T IA,G I A N N E C C H I N IG,C A R I E L L O A,e t a l.S t e n o s i so ft h el a c h r y m a ls y s t e m i n R u b i n s t e i n-T a y b is y n-d r o m e[J].O p h t h a l m o l o g i c aJ o u r n a l I n te r n a t i o n a ld O p h t a l-m o l o g i eI n t e r n a t i o n a lJ o u r n a lo f O p h t h a l m o l o g y Z e i t s c h r i f tF u rA u g e n h e i l k u n d e,2002,216(4):272-276.[10]B A R T S C H O,K R E S S W,K E M P F O,e t a l.I n h e r i t a n c e a n dv a r i a b l e e x p r e s s i o n i nR u b i n s t e i n-T a y b i s y n d r o m e[J].A m e r i-c a nJ o u r n a l o fM ed i c a lGe n e t i c sP a r tA,2010,152A(9):2254-2261.[11]A L-Q A T T A N M M,R A H B E E N I ZA,A L-H A S S N A NZN,e t a l.C h r o m o s o m e16p13.3c o n t i g u o u s g e n e d e l e t i o n s y n d r o m e i n c l u d i n g t h e S L X4,D N A S E1,T R A P1,a n d C R E B B Pg e n e s p r e s e n t i n g a sar e l a t i v e l y m i l d R u b i n s t e i n-T a y b is y n-d r o me p h e n o t y p e:a c a s e r e p o r tof aS a u d ib o y[J].C a s eR e-p o r t s i nG e n e t i c s,2020,2020:6143050.[12]B A R T S C H O,S C HM I D TS,R I C H T E R M,e t a l.D N As e-q u e n c i n g o f C R E B B P d e m o n s t r a t e sm u t a t i o n s i n56%o f p a-t i e n t sw i t hR u b i n s t e i n-T a y b i s y n d r o m e(R S T S)a n d i n a n o t h e r p a t i e n tw i t h i n c o m p l e t eR S T S[J].H u m a nG e n e t i c s,2005,117(5):485-493.[13]B A R T S C H O,R A S I S,D E L I C A D O A,e t a l.E v i d e n c e f o r an e wc o n t i g u o u s g e n e s y n d r o m e,t h ec h r o m o s o m e16p13.3d e-l e t i o n s y n d r o m ea l i a ss e v e r e R u b i n s t e i n-T a y b is y n d r o m e[J].H u m a nG e n e t i c s,2006,120(2):179-186.[14]PÉR E Z-G R I J A L B A V,G A R CÍA-O G U I Z A A,LÓP E Z M,e ta l.N e wi n s i g h t s i n t o g e n e t i cv a r i a n ts p e c t r u m a n d g e n o t y p e-p h e n o t y p ec o r r e l a t i o n so f R u b i n s t e i n-T a y b is y n d r o m ei n39C R E B B P-p o s i t i v e p a t i e n t s[J].M o l e c u l a rG e n e t i c s&G e n o m i cM e d i c i n e,2019,7(11):e972.[15]R U S C O N ID,N E G R IG,C O L A P I E T R O P,e t a l.C h a r a c t e-r i z a t i o no f14n o v e l d e l e t i o n s u n d e r l y i n g R u b i n s t e i n-T a y b i s y n-d r o m e:a nu p d a te of t h eC R E B B Pd e l e t i o nr e p e r t o i r e[J].H u-m a nG e n e t i c s,2015,134(6):613-626.[16]S P E N AS,M I L A N ID,R U S C O N ID,e t a l.I n s i g h t s i n t o g e-n o t y p e-p h e n o t y p ec o r r e l a t i o n sf r o m C R E B B P p o i n t m u t a t i o n s c r e e n i n g i n a c o h o r t o f46R u b i n s t e i n-T a y b i s y n d r o m e p a t i e n t s [J].C l i n i c a lG e n e t i c s,2015,88(5):431-440.[17]Y A O TP,O HSP,F U C H SM,e t a l.G e n e d o s a g e-d e p e n d e n te m b r y o n i cd e v e l o p m e n ta n d p r o l if e r a t i o nd e f e c t s i n m i c e l a c-k i n g t h e t r a n s c r i p t i o n a l i n t e g r a t o r p300[J].C e l l,1998,93(3): 361-372.[18]O I K EY,HA T A A,MAM I Y A T,e t a l.T r u n c a t e dC B P p r o-t e i n l e a d s t o c l a s s i c a l R u b i n s t e i n-t a y b i s y n d r o m e p h e n o t y p e s i n m i c e:i m p l i c a t i o n sf o ra d o m i n a n t-n e g a t i v e m e c h a n i s m[J].H u m a n M o l e c u l a rG e n e t i c s,1999,8(3):387-396.[19]K A L K HO V E N E.C B Pa n d p300:H A T s f o rd i f f e r e n to c c a-s i o n s[J].B i o c h e m i c a l P h a r m a c o l o g y,2004,68(6):1145-1155.[20]C H A N H M,L A T HA N G U E N B.p300/C B P p r o t e i n s:H A T s f o r t r a n s c r i p t i o n a l b r i d g e sa n ds c a f f o l d s[J].J o u r n a l o fC e l l S c i e n c e,2001,114(P t13):2363-2373.[21]S H E I K H B N,A K H T A R A.T h em a n y l i v e so fK A T s d e-t e c t o r s,i n t e g r a t o r s a n dm o d u l a t o r s o f t h e c e l l u l a r e n v i r o n m e n t [J].N a t u r eR e v i e w sG e n e t i c s,2019,20(1):7-23. [22]LÓP E Z M,S E I D E LV,S A N T I BÁÑE ZP,e t a l.F i r s t c a s e r e-p o r t o f i n h e r i t e dR u b i n s t e i n-T a y b i s y n d r o m e a s s o c i a t e dw i t ha n o v e l E P300v a r i a n t[J].B M C M e d i c a lG e n e t i c s,2016,17(1):97.[23]P A R KE,K I M Y,R Y U H,e t a l.E p i g e n e t i cm e c h a n i s m so fR u b i n s t e i n-T a y b is y n d r o m e[J].N e u r o M o l e c u l a r M e d i c i n e, 2014,16(1):16-24.[24]A L A R CÓNJM,MA L L E R E TG,T O U Z A N IK,e t a l.C h r o-m a t i na c e t y l a t i o n,m e m o r y,a n dL T Pa r e i m p a i r e d i nC B P+/-m i c e:a m o d e lf o rt h ec o g n i t i v ed e f i c i ti n R u b i n s t e i n-T a y b i s y n d r o m e a n d i t s a m e l i o r a t i o n[J].N e u r o n,2004,42(6):947-959.(本文编辑马伟平)Copyright©博看网. All Rights Reserved.。
1例携带NIPBL基因新变异的德朗热综合征
98南昌大学学报(医学版)2021年第 61卷第2期Journal of Nanchang University(Medical Sciences)2021,Vol.61No.2 1例携带NIPBL基因新变异的德朗热综合征谢芸芸",袁宇峰",李维君b,李守明a(江西省儿童医院a.儿童保健科;b.临床营养科,南昌330006)摘要:目的明确1例特殊面容、生长迟缓、肢体异常患儿的致病基因变异及来源,为疾病的诊断和遗传咨询提供依据.方法采集患儿及其父母外周血样并提取DNA,进行全外显子组测序检测,结合生物信息学分析方法进行数据分析,获取致病性基因变异位点,最后利用Sanger测序进行验证.结果全外显子组测序结果显示位于5号染色体的NIPBL基因42号外显子上发现错义变异c.7177T>C(p.Ser2393Pro),该变异各人群数据库中未见收录.经父母Sanger验证显示,父母均无携带,属于新发变异(de novo mutation).结合临床症状和基因检测结果,该患儿确诊为经典型德朗热综合征(Cornelia de Lange syndrome,CdLS).结论对于特殊面容、生长迟缓、肢体异常的患儿应考虑CdLS可能,并常规进行基因检测明确诊断,有助于患儿随访管理、预后评估及遗传咨询等.关键词:德朗热综合征;NIPBL;新变异;婴儿;病例报告中图分类号:R725.9文献标志码:A文章编号:20954727(2021)02—0098—04DOI:10.13764/ki.ncdm.2021.02.021A Case of Cornelia de Lange Syndrome Harboring aNovel Mutation of NIPBLXIE Yun-yun a,YUAN Yu-feng a,LI Wei-jun b,LI Shou-ming a(a.DepartmentofChildHealthcare;b.DepartmentofClinicalNutrition,Jiangxi Provincial Children Hospital Nanchang330006,China)ABSTRACT:Objective To identify the mutation and origin of causal genes in a child with unusual facies?growth retardation and limb abnormalities?and to provide a basis for diagnosis and genetic counseling of the disease.Methods Peripheral blood samples were collected from the child and his parents for the extraction of DNA.Whole-exome sequencing and bioinformatics analysis were performed to identify the pathogenic gene mutations.Finally?Sanger sequencing was used to verify the results.Results Whole-exome sequencing showed a missense mutation(c.7177T>C, p.Ser2393Pro)in exon42of the NIPBL gene on chromosome5.The mutation was not included in all population databases.Sanger sequencing indicated that the mutation was a de novo mutation, which was not derived from his parents.Clinical symptoms combined with genetic testing results confirmed that the child had a classic Cornelia de Lange syndrome(CdLS).Conclusion CdLS should be considered in children with unusual facies,growth retardation and limb abnormalities.Furthermore,genetic testing should be routinely performed to confirm the diagnosis.I is helpful for the follow-up management,prognosis assessment and genetic counseling.收稿日期:2020-0806基金项目:江西省卫健委普通科技计划(SKJP2020202159)作者简介:谢芸芸(1987—),女,硕士,主治医师,主要从事儿童保健、营养喂养、食物过敏的研究.通信作者:李守明,主任医师,E-mail:138****************.谢芸芸等:例携带NIPBL基因新变异的德朗热综合征99 KEY WORDS:Cornelia de Lange syndrome;NIPBL;novel mutation;baby;case report德朗热综合征(Cornelia de Lange syndrome, CdLS)(OMIM井122470,300590,300882,610759, 614701)是1933年由荷兰儿科医生Cornelia de Lange最先报道,它是一种多器官受累的先天遗传罕见性疾病,活产婴儿中其发病率为1/10000~ 30000[]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Re s u l t s: A d e l e t i on mut a t i o n( c .5 7 9 0d e l C)wa s f oun d i n t he CREB— bi n di n g pr o t e i n g e ne, CREBBP , whi c h i s r e s p on s i b l e f o r t he 5 0 t o 7 O o f t h e RSTS p a t i e nt s . Thi s mu t a t i on wou l d c a us e a f r a me s h i f t
Pol y me r a s e c h a i n r e a c t i on f ol l o we d by Sa ng e r s e qu e nc i c a nd i d a t e v a r i a n t s i n
中国科学杂志社北京大学学报自然科学版编辑部软件学报编辑部等也都与作者签署相关的著作权转让书中国高校自然科学学报研究会还在会员单位内推广这一工作建议作者将论文的汇编权复制权发行权信息网络传播权翻译权等在全世界范围内转让给期刊社编辑部和相关数据库
・
55 2 ・
生 殖 医学 杂 志 2 0 1 7年 6月 第 2 6 卷第 6 期
wi t h RS TS a f t e r c a r e f ul l y phy s i c a l e x a mi na t i o n. Li ke l y p a t ho g e n i c v a r i a nt s we r e f i l t e r e d a nd s e l e c t e d .
DOI : 1 0 . 3 9 6 9 / j . i s s n . 1 0 0 4 — 3 8 4 5 . 2 0 1 7 . 0 6 . 0 0 9
C REBBP 基 因 突 变 所 致 Ru b i n s t e i n — Ta y b i 综 合 征 1例
罗敏娜 ,曹 宗富 ,王琳 ,张艳 萍 ,魏 莹 ,高华 方 ,陈剑 虹 ,马旭 H
CH EN Ji a n — h o n g . M A Xu
1 . Na t i o n a l Re s e a r c h I n s t i t u t e f o r Fa mi l y Pl a n n i n g, Be i j i n g 1 0 O 0 8 1
a n d Ch i l d r e n ' s Ho s p i t a 1 . Th e wh o l e e x o me s e q u e n c i n g wa s a p p l i e d t o e x a mi n e t h e DNA s a mp l e o f t h e g i r l
CR EBBP g e n e m ut a t i o n i n a pa t i e nt wi t h Ru b i ns t e i n — Ta y b i s y n d r o me
LUO Mi n — n a , C AO Zo n g — f u ,WANG Li n , ZHANG Ya h — pi n g , WEI " k i n g , GAO Hu a — f a n g ,
C RE B B P基因的 c . 5 7 9 0 d e l c位 点 为 引 起 该 R u b i n s t e i n - T a y b i 综 合 征 的突 变 位 点 。 【 关键词1 R u b i n s t e i n - Ta y b i 综合征 ; C RE B B P基因 ; 全外显子组测 序 ; 发育迟缓 ; 宽拇指( 趾)
( RSTS)a nd i d e nt i f y t he ge ne t i c c h a r a c t e r s . Me t ho ds : The c l i ni c a l a na l y s i s wa s p e r f o r me d f o r a p a t i e n t wi t h RSTS i n t he Fi r s t H ui z ho u W ome n
( 1 .国 家卫 生计 生 委科 学 技 术 研 究 所 , 北京 1 0 0 0 8 1 ; 2 .惠 州 第 一 妇 幼 保 健 院 , 惠州 5 1 6 0 0 1 )
【 摘要】 目的 分 析 1例 R u b i n s t e i n - Ta y b i 综合 征的临床及遗传学特 点 。 方 法 对 惠 州 第 一 妇 幼 保 健 院 疑 似 Ru b i n — s t e i n — Ta y b i 综 合 征病 例 进 行 临 床 分 析 , 同 时提 取 先 证 者 D NA 进 行 全 外 显 子 组 测 序 , 筛选致病突变位点 , 用 S a n g e r 测 序 对 突 变 位 点进行验证 。 结 果 在 患 儿 的 1 6 号 染色体 C RE B B P 基 因 上 存 在 1个 杂 合 突 变 位 点 c . 5 7 9 0 d e l c, 这 个 位 点 的 缺 失 造 成 了 蛋 白质 翻 译 的 提 前 终 止 。该 位 点 在 其 父 母 中 均未 发 生 突 变 , 为d e n O V O突变 。 结论 全 外 显 子组 测 序 结 合 S a n g e r 测序 发 现
2 .The Fi r s t H ui z ho u Wo me n and Chi l dr e n' s Ho s pi t al , Hu i z ho u 5 1 6 0 01
[ Ab s t r a c t l
Ob j e c t i v e : To a n a l y z e t h e c l i n i c a l p h e n o t y p e o f a 4 - y e a r s — o l d g i r l wi t h Ru b i n s t e i n — Ta y b i s y n d r o me