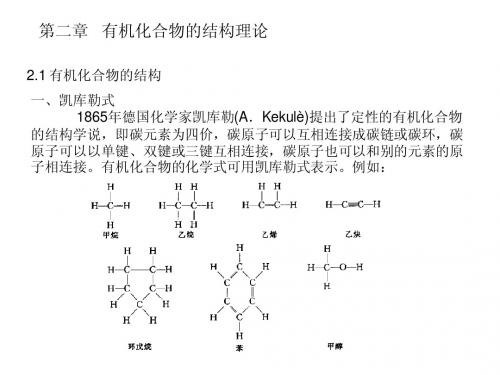
有机化合物结构理论
有机化合物的结构和性质

洪德规则(Hund's rule) 洪德规则
洪德在总结大量光谱和电离势数据的基础上 提出:电子在简并轨道上排布时,将尽可能分 占不同的轨道,且自旋平行[5]。对于同一个电 子亚层,当电子排布处于 全满(s^2、p^6、d^10、f^14) 半满(s^1、p^3、d^5、f^7) 全空(s^0、p^0、d^0、f^0) 时比较稳定。
示例 H:1s^1 F:1s^2∣2s^2,2p^5 ∣ S:1s^2∣2s^2,2p^6∣3s^2,3p^4 ∣ ∣ Cr: 1s^2∣2s^2,2p^6∣3s^2,3p^6,3d^5∣4s^1(注 ∣ ∣ ∣ 意加粗数字,是3d^5,4s^1而不是3d^4,4s^2, 因为d轨道上,5个电子是半充满状态,这里 体现了洪德规则)。
•
1874年范荷夫和勒贝尔建立分子的立体概念, 说明了对映异构和顺反异构现象. • *碳原子总是四价的,碳原子自相结合成键, 构造和构造式 • 分子中原子的连接顺序和方式称为分子的 构造.表示分子中各原子的连接顺序和 • 方式的化学式叫构造式(结构式).用两小点 表示一对共用电子对的构造式叫电子式,用短横 线(-)表示共价键的构造式叫价键式.有时可用只 表达官能团结构特点的化学式,既结构简式.
电子排布
综述
电子在原子轨道的运动遵循三个基本定理: 1、能量最低原理 2、泡利不相容原理 3、洪德定则
能量最低原理 能量最低原理
定义:核外电子在运动时,总是优先占据
能量更低的轨道,使整个体系处于能量最低 的状态。
泡利不相容原理
物理学家泡利在总结了众多事实的基础上提出 :不可能有完全相同的两个费米子同时拥有样的 量子物理态。泡利不相容原理应用在电子排布上 ,可表述为:同一轨道上最多容纳两个自旋相反 的电子。该原理有三个推论 ①若两电子处于同一轨道,其自旋方向一定不 同; ②若两个电子自旋相同,它们一定不在同一 轨道; ③每个轨道最多容纳两个电子。
第二章 有机化合物的结构理论
H—H键的电子云是围绕键轴对称分布的,这种类型的键叫做σ键。
2.共价键的饱和性。 如果一个未成对电子已经配对,就不能再与别的原子的未成对电 子配对。例如氯化氢分子中的氢原子和氯原子的未成对电子已互相配 对,就不能再与其它的原子形成共价键。
3.共价键的方向性。 原子轨道互相重叠程度越大,体系能量就越低,形成的共价键也 就越牢固,因而应使原子轨道最大限度地互相重叠。例如两个2px轨道 只有在x轴方向上才能最大限度地互相重叠形成σ键。两个原子的p轨 道若互相平行,则在侧面能有最大的重叠,这种类型的共价键叫做π 键,π电子云分布在两个原子键轴的平面的上方和下方。
2.异裂(heterolytic) 共价键异裂时,成键的一对电子为某一个原子或基团占有,生 成正离子和负离子。通过共价键的异裂的反应叫做离子型反应。 大多数有机反应都是离子型反应或自由基反应。此外还有协同 反应,在协同反应中,既无自由基也无离子生成,共价键的断裂 和形成是同时进行的。
2.3. 分子间的弱相互作用力 一. 分子间的弱相互作用力类型 分子中相连原子之间存在强烈的吸引力,这种吸引力叫做化学键, 它是决定分子化学性质的重要因素。在物质的聚集态中,分子之间还存在 着一种弱的吸引力,把它统称为范德华(van der Waals)引力,它是决定 物质的沸点、熔点、气化热、熔化热、溶解度、粘度、表面张力等物理化 学性质的重要因素。从本质上讲,这种吸引力是由于分子中电荷分布不均 匀[或瞬间分布不均)而出现的静电作用力。常见的有如下3种。 (1). 偶极-偶极作用力(静电力):这种作用力产生于极性分子的静电相互作 用。如氯甲烷分子中,氯原子电负性较大,氯原子一端带有部分负电荷, 而碳原子上带有部分正电荷。一个分子带负电荷的一端吸引另一个分子带 正电荷的一端,于是分子间出现正负极相吸的排列,即
《有机化合物的结构》杂化轨道理论

《有机化合物的结构》杂化轨道理论在探索有机化合物的结构时,杂化轨道理论是一个极其重要的概念。
它为我们理解有机分子的成键方式、几何构型以及化学性质提供了坚实的理论基础。
让我们先来了解一下什么是原子轨道。
原子轨道可以简单地理解为电子在原子核外可能出现的区域。
在未形成化学键时,原子中的电子处于特定的原子轨道,如 s 轨道、p 轨道等。
然而,当原子参与形成化学键时,为了更好地重叠形成稳定的化学键,原子的原有轨道会发生“杂化”。
杂化轨道理论认为,原子在形成分子时,同一原子中能量相近的原子轨道会重新组合,形成一组新的、能量相同、空间取向不同的杂化轨道。
常见的杂化类型有 sp 杂化、sp²杂化和 sp³杂化。
sp 杂化是由一个 s 轨道和一个 p 轨道杂化而成。
例如,在乙炔(C₂H₂)分子中,碳原子就采用了 sp 杂化。
经过杂化后,两个 sp 杂化轨道呈直线形分布,夹角为 180 度。
每个碳原子用一个 sp 杂化轨道与氢原子的 1s 轨道重叠形成碳氢σ 键,两个碳原子之间则用各自的另一个 sp 杂化轨道重叠形成碳碳σ 键,而两个未参与杂化的 p 轨道则两两重叠形成两个π键。
sp²杂化是由一个 s 轨道和两个 p 轨道杂化形成。
以乙烯(C₂H₄)为例,碳原子采用 sp²杂化,三个 sp²杂化轨道在同一平面上,夹角约为 120 度。
每个碳原子用两个 sp²杂化轨道分别与两个氢原子的 1s 轨道形成两个碳氢σ 键,两个碳原子之间用各自的一个 sp²杂化轨道重叠形成一个碳碳σ 键,而未参与杂化的 p 轨道则相互平行重叠形成一个π键。
sp³杂化是由一个 s 轨道和三个 p 轨道杂化而成。
在甲烷(CH₄)分子中,碳原子就进行了 sp³杂化。
四个 sp³杂化轨道呈正四面体分布,夹角为109°28′。
碳原子用四个 sp³杂化轨道分别与四个氢原子的 1s 轨道重叠形成四个碳氢σ 键。
化学简单有机知识点总结

化学简单有机知识点总结一、有机化合物的结构有机化合物的结构由碳元素和氢元素组成,其中碳是有机化合物的主要元素。
除了碳和氢,有机化合物中还可能含有氧、氮、硫、磷等元素。
有机化合物根据碳原子间的连接方式可分为链状、环状和支链状。
根据碳原子的价态可分为饱和碳原子和不饱和碳原子。
根据碳原子的空间构型可分为手性和非手性化合物。
有机化合物的结构可以用分子式、结构式、构象式、键链式等形式表示。
其中,分子式是化学式的简化形式,用元素符号和原子数目表示分子中原子的种类和数量。
结构式是用线条或点来表示原子之间的连接方式和空间结构。
构象式是用空间构象表示有机分子的立体结构。
键链式则利用化学键的方式来表示分子的构成。
二、有机化合物的性质有机化合物的性质无论是物理性质还是化学性质都十分复杂多样。
其中,物理性质包括外观、颜色、气味、溶解度、沸点、熔点等。
有机化合物的物理性质受分子大小、分子形状和相互作用力等因素的影响,表现出较大的差异性。
有机化合物的化学性质主要包括燃烧、氧化还原、加成反应、消除反应、取代反应等。
有机化合物能进行氢化、卤代、硝化、磺化、烷基化、酯化等多种反应。
有机化合物的化学性质的复杂性主要归因于其分子中含有不同种类的功能基团。
三、有机化合物的反应有机化合物的反应是化学合成和分解的过程,是有机化学中的一大重要内容。
有机反应的分类有很多种,主要可以分为加成反应、消除反应、取代反应和重排反应等。
有机反应受到反应条件、催化剂、反应物质结构等因素的影响,表现出较大的复杂性。
例如,加成反应是指两个分子之间发生共价键的形成。
消除反应是指两个或两个以上的原子团或原子之间发生键的断裂。
取代反应是在有机分子中某一原子或原子团与另一种原子或原子团相互替换。
重排反应是指已有的共价键在不断投入该分子中的新原子所代替,新的共价键的形成,使得原子团的位置发生改变。
四、有机化学物质的应用有机化合物在生产生活中有着广泛的应用。
例如,甲烷是天然气的主要成分,可作为燃料供应给工业和生活用火。
有机化合物的结构、命名、同分异构现象及电子效应

(2)σ 键与π 键:
σ键 两个原子的轨道沿键轴方向重叠,电子云绕着键 轴对称分布。
π键
两个原子的轨道互相平行 进行最大的侧面重叠,电 子云分布在两个原子键轴 的平面的上方和下方。
甲烷的分子结构
乙烷的分子结构
乙烯的分子结构
乙炔的分子结构
(3)电子的离域——离域键 分子轨道理论:组成 分子的所有原子的价 电子不只从属于相邻 的原子,而是处于整 个分子的不同能级的 分子轨道中。
N 1 H
N
1
O
1
OCH3
H 3C
N
2- 甲 基 - 4- 甲 氧 基 吡 啶
9、部分化合物的俗名、部分缩写
蚁酸、醋酸、草酸、硬脂酸、软脂酸、酒石 酸、肉桂酸、苦味酸、葡萄糖、果糖、麦芽 糖、蔗糖、核糖、脱氧核糖、甘氨酸、卤仿、 甘油;DMF、THF、DMSO、DNA、RNA
要点:
1、掌握母体的选择、主碳链的选择以 及编号原则。
5 - 溴 - 2 - 己 炔
6、次序规则
在系统命名法中,取代基排列的先后顺序、顺反构 型的确定、手性化合物的构型等都是根据次序规则, 按一定的方法确定。 (i) 将原子或原子团游离价所在的原子按原子序数大 小排列,原子序数大的原子优先于原子序数小的 原子。 例如:Br>Cl>O>C>H (ii) 对同位素元素,则按相对原子质量大的优先于相 对原子质量小的排列。 例如:T>D>H
路易斯式
H H HC C H H H H H HC C H
H H CC HC CH CC H H
凯库勒式
H H HC C H H H
H H C C H H
结构简式:CH3CH2CH2CH2CH3 键线式
第九讲 有机化学结构理论

共价键的键参数
4) 键矩、分子偶极矩 δ+ H δCl 非极性共价键 H-H Cl-Cl 极性共价键 H-Cl
偶极矩()有方向性
H3C
δ + 部分正电荷 δ - 部分负电荷 箭头由正端指向负端、指向 电负性更大的原子。
C C CH3 Br CH3CH2 O CH2CH3
H
Cl
Br
净偶极矩指向 多原子分子的偶极矩,是各键的偶极矩向量和。
芳香性(休克尔规则) 1931 年德国化学家休克尔(Hückel)从分子轨道理 论的角度,对环状多烯烃(亦称轮烯)的芳香性提出
了如下规则,即Hückel规则。 其要点是:化合物是轮烯,共平面, 它的π电子数为4n+2 (n为0,1,2,3…,n整数), 共面的原子均为sp2或sp杂化。
双键与轮烯直接相连,计算电子数时,将双键写成其 共振的电荷结构,负电荷按2个电子计,正电荷按0计, 内部不计。如下面物质均有芳香性:
偶极矩的应用
• 丙烯与卤化氢加成时,主要产物是2-卤丙烷。即 当不对称烯烃与卤化氢加成时,氢原子主要加到 含氢较多的双键碳原子上,这一规律称为马尔科 夫尼可夫规则,简称马氏规则。
偶极矩的应用
•
CH3CH==CHCH3 + CH3CH2CH==CH2
• 在醇脱水或卤代烷脱卤化氢中,如可能生成两种烯 烃的异构体时,则在生成的产物中双键主要位于烷 基取代基较多的位置。这一规律称为扎伊采夫规则。 例如,2-丁醇脱水时,主要产物是2-丁烯,1-丁烯 是次要的。
例如,1-丁烯:
H 3C H
CH2=C
H CH2CH3
H 3C H H
没有顺反异构
CH3 H
CH3
顺-2-丁烯
01有机结构理论.

3 比较酸性大小并说明理由
4 解释:4-硝基苯酚的酸性为 3,5-二甲基-4-硝基苯酚的 40 倍。
5 从空间效应和共振理论两个角度解释下列结果:
6 SN1 反应活性:
7 预测下列两个平衡反应中,哪个更趋向右边?为什么?
8 预测下列反应发生的位置:
9 试解释下列反应的产物是(A)而不是(B):
任何含氢的分子或离子都是一种潜在的B酸,
来自共振作用获得的额外稳定化能(称为共振能)。各共振结构的稳定性越接近,共振能越 大。存在着共振作用的分子(或离子、自由基)比没有共振作用的稳定。 (4) 共振作用以双箭号“↔”表示,它描写的是共振杂化体的各路易斯结构相互叠加的非 平衡关系,不能与化学平衡相混淆。
二 共振理论
2.正确书写共振结构式的规则 (1) 含价键数目较少的或形式电荷分离较大的结构是能量较高的不稳定结构,对 共振杂化体的贡献小。例如:
2. 对映异构 实物与其镜像不能互相重合的性质称为手性或手征性。
具有手性的分子称为手性或手征性分子。
手性分子与其镜像互为对映异构体,简称对映体。 构造相同的分子,由于构型不同而产生互呈镜像,手性不同分子的异构现象称为 对映异构现象; 而产生彼此不呈镜像关系的构型异构体(被称为非对映异构体)的异构现象称为 非对映异构现象。 由于历史上手性和旋光性密切相关,以致手性和旋光性,对映异构和旋光异构互 换 地使用。
下面以 NO2为例子说明路易斯结构式的书写程序: (1) 计算电中性原子的价电子总数、中性分子的总价电子总数等于组成该分子的全部中性 原子的价电子数目之和。若是离子,每一个负电荷使价电子总数增加 1;每一个正电荷使价 电子总数减少 1。NO2的价电子总数等于 17。 (2) 初步写出结构式。 写出各原子实符号,填入由步骤(1)所确定的价电子,在不违反价壳占据度原则 下,使共价键数目最多,未共享价电子数目最少。于是,NO2的结构式初步写为:
有机化合物的结构

有机化合物的结构除了分子式,有机化合物的结构还可以通过分子模型来描述。
分子模型使用球和棒表示原子和键的结构。
原子通常使用彩色小球表示,而连接原子的化学键则使用棒状物表示。
有机化合物的结构包括分子中原子之间的连接方式以及它们在空间中的排列方式。
分子中原子之间的连接通常使用共价键来实现。
共价键是一种通过原子之间的电子共享来保持原子在一起的键。
这种共享可以将原子连接成链、环和分支等不同的结构。
化合物中的共价键可以是单键、双键或三键,它们的强度和长度会有所不同。
有机化合物的结构也涉及键的性质。
共价键可以是极性的或非极性的。
极性键是由于连接原子之间电子的不均匀分布而产生的。
一个极性键可能会由于一个原子吸引更多的电子而带有部分负电荷,而另一个原子可能会带有部分正电荷。
这种区分正负电荷的分布对于有机化合物的反应和性质具有重要影响。
另一个重要的结构概念是立体化学。
立体化学描述了有机化合物中原子或基团在空间中的排列方式。
有机化合物可以具有手性和非手性结构。
手性分子是在镜面上不对称的分子,它们可以存在两个镜像异构体,称为对映体。
非手性分子是镜面对称的分子,它们没有对映体。
手性分子和非手性分子可以具有不同的化学性质,并且在生物学和药学领域中具有重要的应用。
至此所述,有机化合物的结构是通过分子式和分子模型来描述的。
分子式提供了关于元素的数量和种类的信息,而分子模型则展示了原子之间的连接方式和在空间中的排列方式。
这些结构信息对于理解有机化合物的性质和反应机理至关重要,也对于合成有机化合物和设计新药物具有重要意义。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第二章有机化合物的结构理论从有机化合物的分类方法中,可以看出同分异构现象在有机化学中占有相当重要的地位。
同分异构体有相同数目相同种类的原子,但原子间连接的次序和空间取向不同,即结构上的不同使分子式一样的化合物包含着不同的化合物组成。
因此,有机化学的学习研究必须从结构上着手才能抓住本质而不致误入歧途。
结构问题如得不到正确认识和解决,那就像Wohler在1835年所说的:“有机化学是充满最特殊事物的热带丛林,却又是一个恐怖的无边际的丛林,无人敢进去,因为相像的找不到出路。
”不解决结构问题,就不可能学习研究有机化学和有机化合物本身。
1857年,Kekule指出每一种原子都有一定的化合力,这种化合力就是价,碳原子的价为四价。
可以说有机化学的结构学说就是在此基础是发展起来的。
1913年,Bohr提出的原子结构理论,产生了原子价的电子学说,标志着经典的结构理论已经过渡到结构的电子理论了。
形成分子的驱动力是因为分子比原子稳定,原子形成分子后能量得到释放。
分子中化学键的形成使体系能量降低,而化学键的断裂总是需要吸收能量。
但是,原子又是如何结合起来才形成分子的呢?要正确回答这个问题就比较困难了。
1917年Kossel和Lewis分别提出,化学键由电子组成,可分为离子键和共价键两大类。
反应时,原子将失去或得到电子,使结构接近惰性气体的结构。
化学变化仅仅涉及核外的电子即价电子的反应,表示键的短线即是一对成对电子,这些观点已经成为现代价键理论的基础。
1926年,Schrodinger等提出了说明原子结构中的电子运动的量子力学理论,而绝大多数化学家都运用了Schrodinger的波动方程理论,使我们对有机化合物结构问题的探索和了解也具有了现代量子理论基础。
2.1 原子轨道描述原子中单个电子运动状态的波函数叫做原子轨道(atomic orbital)。
例如氢原子,若将原子核定为坐标原点,则单个电子在空间运动状态可由正坐标系x,y,z或球极坐标系r,θ,φ来确定。
那么,描述该电子在空间运动状态的波函数,即原子轨道,可用φ(x,y,z)或φ(r,θ,φ)来表示。
Schrodinger方程是表述微观物体运动的方程。
用Schrodinger方程求解氢原子中电子运动状态时,得到主量子数n,副量子数l,磁量子数m三个量子数,它们之间的关系为:主量子数n是用来描述原子中电子出现几率最大区域离核的远近,或者说它是决定电子层数的,主量子数n的取值为1,2,3…等正整数。
n=1代表电子离核的平均距离最近的一层,即第一电子层;n=2代表电子离核的平均距离比第一层稍远的一层,即第二电子层。
副量子数l又称角量子数。
当n给定时,l可取值为0,1,2,3…(n-1)。
在每一个主量子数n中,有n个副量子数,其最大值为n-1。
副量子数l的物理意义之一是表示原子轨道(或电子云)的形状,其二是表示同一电子层中具有不同状态的亚层。
例如,n=3时,l 可取值为0,1,2。
即在第三层电子层上有三个亚层,分别为s,p,d亚层。
为了区别不同电子层上的亚层,在亚层符号前面冠以电子层数。
例如,2s是第二电子层上的亚层,3p是第三电子层上的p亚层。
表2.1列出了主量子数n,副量子数l及相应电子层、亚层之间的关系。
表2.1 主量子数n,副量子数l及相应电子层、亚层之间的关系磁量子数m决定原子轨道(或电子云)在空间的伸展方向。
当l给定时,m的取值为从-l到+l之间的一切整数(包括0在内),即0,±1,±2,±3,…±l,共有2l+1个取值。
即原子轨道(或电子云)在空间有2l+1个伸展方向。
三个量子数的每一种组合代表电子的一种运动状态,即一个原子轨道。
例如,l=0时,m只能有一个值,即m=0,说明s亚层只有一个轨道为s轨道,该轨道电子云呈球形对称分布,没有方向性。
当l=1时,m可有-1,0,+1三个取值,说明p电子云在空间有三种取向,即p亚层中有三个以x,y,z轴为对称轴的px,py,pz轨道。
当 l=2时,m可有五个取值,即d电子云在空间有五种取向,d亚层中有五个不同伸展方向的d轨道。
各种常见原子轨道的形状与空间取向如下图所示:在有机化学中,了解碳原子的原子轨道状态以及成键方式是极为重要的。
碳原子的原子序数为6,原子核外存在6个电子,它们分别填充在1s、2s、和2p轨道上,其电子构型为1s22s22p x12p y12p z,其中1s轨道属于内层原子轨道,其中的两个电子一般认为不参与成键。
2.2 共价键理论2.2.1 路易斯(Lewis)理论1916年,美国的 Lewis 提出了共价键理论。
认为分子中的原子都有形成稀有气体电子结构的趋势,而达到这种稳定结构,并非通过电子转移形成离子键来完成,而是通过共用电子对来实现。
这种理论很好地解释了氢原子通过共用一对电子,使每个H均成为He 的电子构型,由此形成稳定的氢气分子。
Lewis的贡献在于提出了一种不同于离子键的新的成键方式, 解释了电负性差别比较小的元素之间原子的成键事实。
但Lewis没有说明这种键的实质, 适应性不强,特别是难以解释BH3, BCl3等未达到稀有气体结构的分子的成键方式。
1927年, Heitler 和 London 用量子力学处理氢气分子, 解决了两个氢原子之间化学键的本质问题,使共价键理论从经典的Lewis理论发展到了目前的现代共价键理论。
一、共价键的形成价键法又称为电子配对法,其主要内容归纳如下:(1)假如原子A与原子B各拥有一个未成对的电子,且自旋方向相反,那么它们就可以互相配对,形成共价(单)键。
如果A和B各拥有两个或三个未成对电子,那么配对形成的共价键就是双键或叁键。
例如,H有一个未成对的1s电子,F有一个未成对的2p电子,它们可以配对构成H-F 单键;N原子含有三个未成对的2p电子,因此相互间可以形成共价N≡N叁键;He原子没有未成对的电子,两个He原子互相接近时,不能形成共价键。
(2)如果A原子有两个未成对电子,B原子有一个未成对电子,那么一个A原子可以与两个B原子相结合。
例如,氧原子拥有两个未成对的2p电子,氢有一个单电子,所以一个氧可以和两个氢结合形成H2O。
因此,原子内含有的未成对的价电子数,通常就是它的原子价数。
(3)一个电子与另一个自旋相反的电子配对以后,就不能再与第三个电子配对。
例如,氢原子拥有一未成对的电子,它们能配对形成H2分子,后者与第三个H原子接近,就不能再结合成为H3分子。
这说明共价键的形成具有饱和性。
(4)两个电子配对也就是它们的原子轨道的重叠。
原子轨道重叠越多,形成的共价键越强。
因此,原子轨道要尽可能在电子云密度最大的方向叠加,即共价键的形成具有方向性。
氢原子的1s轨道呈球型分布,无方向性,而氟原子的2p轨道呈哑铃型分布,电子云在其对称轴(z轴)上电子云密度最大,即具有方向性。
当氢与氟的原子轨道叠加时,只有沿z轴方向接近时,才能发生最大程度的重叠而形成稳定的共价键,其它方向接近都不能获到最大的重叠。
(5)能量相近的原子轨道,可以进行杂化,组成能量相等的杂化轨道,这样可以使成键能力增强,体系的能量降低,而成键后可达到最稳定的分子状态。
具体方式在下一节中详述。
二、共价键的方向性和饱和性共价键的数目由原子中未成对的电子数来决定(包括原有的和激发而生成的)。
例如,氧原子拥有两个未成对电子,氢原子拥有一个未成对电子, 可以结合形成水分子, 即氧原子只能形成2个共价键。
而碳原子拥有四个未成对的价电子(激发生成),能与四个氢原子形成四个共价单键。
原子中未成对电子的数目决定了共价键的数目,这就是共价键的饱和性。
各原子轨道在空间中具有固定的分布方向, 为了满足轨道的最大重叠, 原子间形成共价键时, 显然具有方向性。
例如,氯的3p z轨道和 H 的1s 轨道重叠, 要沿着z轴重叠, 从而保证最大重叠, 而且不改变原有的对称性。
氯的3p z轨道自身叠加,也需要保持对称性和最大重叠。
下面“头碰头”的重叠方式是有效的,而“头碰腰”的重叠方式是无效的。
三、共价键的键型成键的两个原子核间的连线被称为键轴。
按成键方向与键轴之间的关系,共价键的键型主要分为两种:即σ-键和π-键。
σ-键的特点:将成键轨道,沿键轴方向旋转任意角度,图形及符号均保持不变。
即成键轨道围绕键轴呈圆柱型对称分布。
例如,氢气分子:氟化氢分子:氯气分子:σ-键的成键方式,可以用“头碰头”的形象化表述来描述。
π-键的特点:成键轨道围绕键轴旋转180°时,图形重合,但符号相反,即键的对称性对通过键轴的节面呈现反对称分布。
例如,氮气分子的两个氮原子沿x轴方向形成σ-键(2p x 轨道头碰头叠加)的同时,2p y轨道和2p z轨道则以“肩并肩”的方式进行叠加,形成两个π-键。
四、共价键参数化学键的形成情况,完全可由量子力学的计算来进行定量描述。
不过,人们习惯上仍用几个物理量加以描述,这些物理量被称为共价键的参数。
键能:以共价键结合的分子在气体状态下裂解成原子(或原子团)时所吸收的能量称为该共价键的解离能。
A-B (g) A (g) + B (g)∆H对于双原子分子,共价键的解离能即等于键能;但对于多原子分子,则要注意解离能与键能的区别与联系。
例如,甲烷的四个碳氢建的解离能分别为:键能则为它们的平均值,即为415.3 kJ.mol-1。
键长:成键的两个原子核之间的距离,叫键长,一般以pm为单位。
键长通常与成键原子的性质以及成键类型紧密相关。
一般来说,原子半径小的原子间形成的共价键的键长较短。
对两个相同原子的成键,一般单键键长最长,双键次之,叁键最短。
键长越短,一般键能越高。
另外,键长还与成键原子杂化轨道的状态有关,杂化轨道中s轨道的成分越高,键长越短。
例如,通常C(sp3)―H的键长为110pm,C(sp2)―H的键长为107pm,而C(sp)―H的键长只有106pm。
在不同化合物中,相同键的键长和键能也会有所不同。
例如,甲醇和甲烷中的碳氢键的键长和键能是不同的。
键角:分子中一个原子与另外两个原子形成的两个共价键在空间形成的夹角,被称为键角。
键角的大小与中心原子的性质直接相关,同时还和与中心原子成键的原子的性质有一定的关系。
例如,水分子H-O-H的键角是104.5o,氨分子中H-N-H的键角是107.3o,甲烷分子是四面体结构,H-C-H的键角为109.5o。
与水分子有所不同,H2S分子的H-S-H 的键角为92°,甲醇分子的C-O-H的键角是108.9o。
五、共价键的极性分子中不同原子间形成的共价键,若两个原子吸引电子的能力不同,那么共用电子对会偏向吸引电子能力较强的原子一方,导致吸引电子能力较弱的原子一方相对的显正电性。
这样的共价键叫做极性共价键,简称极性键。