第四章能量色散X光谱EDS
第四章能量色散X光谱EDS培训讲学

(二)获取EDS谱
(1)首先,应尽量利用Ka谱线,因为L和M谱线中重叠 谱峰太多,除非是Ka谱线对Si(Li)探头来说能量太高,比 如原子序数比较高的元素。
(2)在样品不发生漂移或者污染的情况下,获取尽可 能高的特征X射线计数率。
(3)使样品的倾转为零,并且样品的刃边对着EDS探头。
(4)如果样品处于动力学衍射的双束条件,应稍微倾转 样品,使得样品偏离双束条件。
(三)扣除背底
所谓背底是指EDS谱中特征峰下面由韧致辐射造成的 连续强度分布。背底的强度随X射线能量的增加而减小。 双窗口法
在特征X射线峰的两侧取等宽的两个 窗口,两窗口积分值的平均值作为背 底强度。 适用与能量大于2keV的特征X射线峰, 并且样品很薄韧致辐射不被吸收的情况
(四)K因子的确定
K因子不是常数,它随电镜、EDS探测器(包括窗口、 固体收集角、取出角等)、样品厚度和成份、获取EDS 谱的收集条件、EDS谱的分析和处理条件等有密切关系。 只有在以上条件都相同的条件下K因子才是常数。如果 条件改变,则K因子也随之改变。
(3)产生电离空位的壳层用K,L,M等表示,产生电子跃 迁的壳层用α,β等表示。例如:Kα表示从电子从L层跃 迁到K层;Kβ表示电子从M层跃迁到K层;Lα表示电子从 M层跃迁到L层。
(4)L壳层分为三个亚壳层,最外面的亚层的电子跃迁到K层产生 的特征X射线称为Kα1,次外层产生的特征X射线称为Kα2。依此类推。 虽然EDS可以记录所以特征X射线,但一般只表征K,L,M和α,β。
第四章能量色散X光谱EDS
§4.1 绪论
EDS 70年代初问世以来,发展速度很快:
(1)分辨率已达到130eV左右,理论分辨率约 100eV
(2)以前Be窗口元素分析范围为11Na-92U,现 在一般都用有机膜超薄窗口,分析元素可从5B-92U。
能量色散x射线荧光光谱

能量色散x射线荧光光谱能量色散X射线荧光光谱学习资料一、基本原理1. X射线与物质的相互作用- 当一束高能X射线照射到样品上时,会与样品中的原子发生多种相互作用。
其中,光电效应是能量色散X射线荧光光谱(EDXRF)的基础。
在光电效应中,原子中的内层电子吸收X射线光子的能量,克服其结合能而被激发逸出原子,从而在原子内层留下一个空穴。
- 外层电子会跃迁到这个空穴来填补,在这个过程中会释放出具有特定能量的特征X射线。
这个特征X射线的能量等于跃迁前后两个能级的能量差,它是元素的特征标识,不同元素的特征X射线能量不同。
2. 能量色散原理- 在EDXRF中,样品受激发产生的特征X射线进入探测器。
探测器将X射线光子的能量转化为电信号,这个电信号的幅度与X射线光子的能量成正比。
- 通过对电信号进行放大、处理和分析,可以得到X射线的能量分布谱图。
在谱图中,不同能量的特征X射线峰对应着不同的元素,峰的强度与该元素在样品中的含量有关。
二、仪器结构1. X射线源- 是产生激发X射线的部件。
常见的X射线源有放射性同位素源和X射线管。
- 放射性同位素源具有稳定、简单、不需要外部电源等优点,但能量不可调且存在放射性安全问题。
X射线管则可以通过调节管电压和管电流来改变X射线的能量和强度,应用更为广泛。
2. 样品室- 用于放置待分析的样品。
样品室的设计要考虑到对不同类型、形状和大小样品的适应性。
- 有些样品室还配备有样品旋转装置,可以使样品在分析过程中均匀受激,提高分析结果的准确性。
3. 探测器- 是仪器的核心部件之一。
常用的探测器有硅锂探测器(Si(Li))和高纯锗探测器(HPGe)等。
- 硅锂探测器在室温下性能会下降,通常需要在液氮温度下工作,它对轻元素有较好的探测能力。
高纯锗探测器具有较高的能量分辨率,但也需要低温冷却,主要用于对能量分辨率要求较高的分析场合。
4. 信号处理与分析系统- 探测器输出的电信号经过前置放大器、主放大器等放大电路进行放大,然后通过多道脉冲幅度分析器(MCA)将不同幅度(对应不同能量)的脉冲信号进行分类和计数。
能量色散x射线光谱

能量色散x射线光谱能量色散X射线光谱(EDX)是一种物理学技术,用来分析物体表面或里面结构的特征,它能精确地探测物质的显微结构、形态和组成,并可以估计有机物的纯度、含量和配比等。
EDX技术可以用于在压力、温度、湿度和环境污染的条件下的物质的分析,在活性物质的研究中成为重要的方法,也是现代材料分析中一种主流技术。
EDX物理学技术通过X射线波长的变化来分析物质的显微结构和表面形态,其基本原理是将X射线辐照到样品,并观察所产生的X射线能谱。
EDX技术可以检测出特征X射线,将其分解成不同元素,并对其分别进行分析。
EDX相比于其它X射线分析技术,分析速度更快,同时通过检测到的X射线能谱可以计算出材料中每种元素的质量比例,提供出高精确度的分析结果。
EDX技术可用于半导体制造工艺、焊接工艺、生物医学领域、薄膜研究等的分析,还可以用于对金属材料的组织和性能的研究,是材料学研究如硅钢、合金、复合材料等的有效分析工具。
例如,EDX技术可以用于研究表面涂层的位置、厚度和元素成分,以及硅钢和合金材料中的灰分含量,甚至还能发现微量元素的存在。
尽管EDX技术对材料分析非常精确,但它也存在一定的局限性,如谱线宽度较宽,分辨能力较低,分析范围有限等。
EDX分析的结果只能反映常温下的样品的特性,而不能反映高温时的特性,所以EDX分析的结果只能用于参考,而不能作为唯一的判断依据。
此外,EDX技术也依赖于较好的技术人员和硬件设备,要求使用者必须具备良好的化学知识和安全措施等技能,才能进行有效的EDX 技术研究和应用。
总之,EDX技术是一种重要的物理分析技术,它可以用于精确检测物质的表面结构、形态和组成,也可以估计有机物的纯度和含量,是很多领域的材料分析的首选手段,但也有一定的局限性,也需要良好的技术人员和硬件设备才能做出准确的结论。
eds能谱表

EDS能谱表一、引言随着科技的不断进步,能谱分析技术已成为材料科学、生命科学、环境科学等领域中不可或缺的分析手段。
其中,EDS能谱表作为一种常用的能谱分析技术,具有广泛的应用前景。
本文将对EDS能谱表的基本原理、技术特点、应用领域及未来发展方向进行详细阐述。
二、EDS能谱表基本原理EDS能谱表,即能量色散X射线光谱仪,是一种基于X射线照射样品后产生的特征X射线来进行元素分析的仪器。
当X射线照射到样品上时,样品中的元素会发射出具有特定波长和能量的特征X射线。
通过测量这些特征X射线的能量和强度,可以确定样品中元素的种类和含量。
EDS能谱表的原理基于X射线与物质相互作用时的能量损失和光谱线特征,能够对样品进行定性和定量分析。
三、EDS能谱表技术特点EDS能谱表具有以下技术特点:1.高精度元素分析:EDS能谱表可以对样品中的元素进行高精度分析,检测范围广泛,包括轻元素到重元素。
2.快速分析:EDS能谱表具有较高的分析速度,可以在较短的时间内完成样品的元素分析。
3.空间分辨率高:EDS能谱表的空间分辨率较高,能够提供元素在样品表面分布的信息。
4.无需样品制备:EDS能谱表分析时不需要对样品进行特殊制备,可以直接对样品进行测量。
5.操作简便:EDS能谱表的操作系统较为简单,便于用户快速掌握。
6.适用范围广:EDS能谱表适用于各种材料的分析,如金属、陶瓷、塑料、生物组织等。
四、EDS能谱表应用领域EDS能谱表在多个领域中都有广泛的应用:1.材料科学:在材料科学领域中,EDS能谱表常被用于合金、陶瓷、复合材料等材料的元素分析和成分研究。
通过对材料表面元素的分布进行分析,可以深入了解材料的结构和性能。
2.生物学:在生物学领域中,EDS能谱表常被用于生物组织、细胞、蛋白质等样品的元素分析。
通过对生物样品中元素的种类和含量进行分析,可以揭示生物体内的代谢过程和生理机制。
3.环境科学:在环境科学领域中,EDS能谱表常被用于土壤、水、空气等样品的元素分析。
X射线能谱(EDS)分析

逃逸峰、合峰 特征X射线(能量Ex)入射到Si(Li)半导体探测 器时,使Si放出K特征X射线(1.740keV),得 到能量为Eesc=Ex-1.740keV的特征X射线 — 逃逸峰 两个特征X射线光子几乎同时进入探测器,探 测器无法进行识别,在两个特征X射线光子的 能量值和位置产生一个峰 — 合峰
五、定量分析中需注意的问题
试样对X射线的吸收
(1)从理论上对k 因子进行修正 (2)利用外插法将实验中获得的不同厚度区 域的测定值外推到很薄的情况,求出理想的 k 因子
统计误差
分析含有的微量元素时,需进行长时间测量ቤተ መጻሕፍቲ ባይዱ 以获得足够的X射线强度,减小误差 标准偏差=N1/2 误差=(N1/2/N)100=N-1/2 100(%)
多道脉冲高度分析器:
X射线能量不同,其电脉冲幅值不同,将 不同幅值的电压脉冲按能量大小分类并 统计,并输出给计算机
三、EDS的分析技术
X射线的测量:连续X射线(部分电子经多次和试样 碰撞产生电磁波)和从试样架产生的散射进入探测器, 形成背底,使用铍制试样架可减小背底,支持试样的 栅网应采用与分析对象的元素不同的材料制作 元素的面分布分析方法:使用扫描像观察装置,使电 子束在试样上做二维扫描,测量特征X射线的强度, 使与这个强度对应的亮度变化与扫描信号同步在阴极 射线管记录部分显示,使用场发射电子枪,能够得到 优于1nm分辨率的元素面分布像
硅铝氧氮耐热陶瓷的X射线元素面分布像
X射线元素线分布图
四、定量分析(薄试样)
定量分析公式:CA/CB=kAB(NA/NB) kAB — k 因子,依赖于物质和装置 CA,CB — A,B元素的浓度(质量分数) NA,NB — A,B元素的特征X射线强度 定量分析实验 k 因子的确定:(1)理论计算(误差较大);(2) 实验确定(精度高,需要组成与被测化合物相近,并 已知组成的标准试样) 对于特征X射线谱重叠的情况,必须根据标准试样 谱进行重叠峰分离后,进行定量分析
电子能谱分析edx
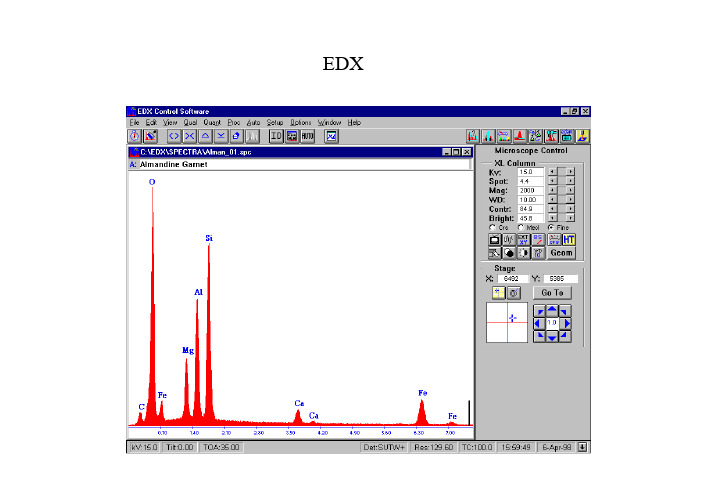
EDXX射线能谱一介绍EDS(energy dispersive spectro-scopy)能量色散谱EDX (Energy Dispersive X-ray)WDS(wavelength dispersive spectro-scopy)波长色散谱EDX:X射线强度和能量曲线,定量分析样品的化学成份主要用途:•1) 非均匀样品的局部化学成份•2) 较少量材料或小颗粒材料的化学成份•3) 非均匀样品种一维或二维的成份分布•4) 沉积在任意衬底上的薄膜成份特点1)铍以上元素2)最小能探测到的重量比:0.1 wt% ——1 wt%3)定量结果的相对误差:2-20%(取决于校正方法等)4)在计算机控制下,1分钟以内可分析16种元素5)空间分辨率取决于平均原子序数、样品密度、束能量等(SEM中0.2——10微米)获得可靠的分析结果要求样品:·1) 样品平整光滑(尤其对定量分析,样品要抛光)2) 可以分析表面粗糙的样品,但仅限于定性和半定量分析3) 样品必须导热导电,必要的时候表面需要喷炭或金推荐书目:Scanning Electron Microscopy and X-ray microanalysisNew York 1992 (生物学、材料科学、地质学)Scanning Electron Microscopy,X-ray microanalysisand Anlytical Electron MicroscopyNew York 1990二定量分析Fig.2: Schematic diagram showingwhere 29<Z<37.Detected Energy (E)Fig.5: Schematic diagram of the intensity variation of the continuum backgroundwith energy, showing the generated and detected background energy.Intensity (I)Generated A )背底和特征峰(二)影响X 射线强度的几种因素B)原子序数对X射线强度的影响Variation in fluorescence yield with atomic number.C )荧光产生率E )Mass absorption coefficient of Fe, for X-rays of varying energyD)X 射线的吸收探测角度:角度越小,X射线吸收越强。
第四章 能量色散X光谱EDS
§4.1 绪论
3 Yuxi Chen Hunan Univ.
EDS 70年代初问世以来,发展速度很快:
(1)分辨率已达到130eV左右,理论分辨率约100eV
(2)以前Be窗口元素分析范围为11Na-92U,现在一 般都用有机膜超薄窗口,分析元素可从5B-92U。
(3)较完善的元素定性、定量分析等软件,定量准 确度也有很大提高,中等原子序数元素基本无重叠峰, 定量准确度已接近于波谱仪。来自CA IA K AB CB IB
这里KAB称为Cliff-Lorimer K因子,K因子不是常数。 K因子与加速电压、样品成份、厚度、EDS探测器的效率 等因素有密切关系。
17 Yuxi Chen Hunan Univ.
(1)在两种元素的情况下,CA+CB =100% (2)在多种元素的情况下,
探测器输出的电压脉冲高度,由电子-空穴对的数量 N决定,形成EDS谱的横坐标-能量。 前臵放大器放大的信号,进入多道脉冲高度分析器, 把不同能量的X射线光子区分开来,在EDS谱上形成 33 Yuxi Chen 纵坐标-脉冲数。 Hunan Univ.
原子内壳层电子电离过程中产生特征X射线的几率。
特征X射线的荧光产额ωk与俄歇电子产额ak的关系为 Ωk + ak =1 例如 Al(Z=13),其ωk≈0.04;ak≈0.96 C(Z=6), 其ωk≈0.001;ak≈0.999
意味着激发1000个C原子才能产生一个特征X光信号。
12 Yuxi Chen Hunan Univ.
14 Yuxi Chen Hunan Univ.
韧致辐射(连续X光)
入射电子穿越原子外壳层与原子核产生非弹性碰撞, 与原子库伦场的交互作用使电子损失一定能量,损失 的能量会以X光的形式释放出来。由于损失的能量取 决于非弹性碰撞的大小,所以X光的能量是连续的, 甚至可以高达入射电子的能量。这种连续的X光称为 韧致辐射。 韧致辐射的强度取决于样品中平均的原子序数Z。在 EDS谱中形成连续的背底,而特征X射线形成一个个不 连续的窄峰,是EDS成份分析的基础。
eds表面成分
"EDS"通常指的是能量色散X射线光谱分析(Energy Dispersive X-ray Spectroscopy),这是一种用于材料分析的技术,通常在扫描电子显微镜(SEM)中使用。
EDS能够提供样品表面或内部元素的化学成分信息,因为不同的元素在受到高能电子束轰击时会发出特征的能量色散X射线。
EDS分析的主要步骤包括:
1. 样品准备:将样品放置在SEM的样品台上,并调整样品的位置,使其在电子束的轰击下可以发出X射线。
2. 电子束轰击:SEM的电子枪发射高能电子束,照射到样品表面。
3. X射线产生:样品中的元素受到电子束的轰击后,会失去电子并发射出特征的能量色散X射线。
4. X射线检测:这些X射线被探测器检测到,并转换为电信号。
5. 数据处理:电信号经过放大和处理后,被转换为谱图,谱图显示了不同元素的特征X射线强度。
6. 谱图分析:通过对照已知元素的X射线谱图,可以确定样品中的元素成分和含量。
EDS分析可以提供有关样品表面化学成分的详细信息,这对于材料科学、地质学、生物医学和其他领域的研究和质量控制非常重要。
然而,EDS分析通常需要专业的设备和培训,以确保准确解释谱图数据。
第四章能量色散X光谱EDS
X射线荧光产额ωk与原子序数的关系
随原子序数Z的增加,特征X射线的荧光产额ωk也增加。
(1)临界电离能量EC(Critical Ionization Energy) 激发原子内壳层电子使原子电离的最小能量。 EC随原子序数增减而增加。 (2)电离截面σ(Ionization Cross Section) 单位面积内的电离数目。电离截面是电离事件发生几率 的数学描述。 在透射电镜中,主要考虑过压比Overvoltage U,也就是 电子束能量和临界电离能Ec的比值。一般Ec<20keV,而 入射电子能量≥100keV, 所以U≥5,电离截面一般是个 常数。如果入射电子能量接近或者稍大于临界电离能, 则电离几率很小。
(四)K因子的确定
K因子不是常数,它随电镜、EDS探测器(包括窗口、 固体收集角、取出角等)、样品厚度和成份、获取EDS 谱的收集条件、EDS谱的分析和处理条件等有密切关系。 只有在以上条件都相同的条件下K因子才是常数。如果 条件改变,则K因子也随之改变。
两种主要的确定K因子的方法 (a)利用标准样品确定K因子; (b)利用第一原理计算
利用标准样品确定K因子
所谓标准样品,就是成份已知,且样品厚度也知道的 TEM样品。 利用标准样品,测得IA和IB的值,由于成份CA和CB已知 则利用Cilff-Lorimer公式就可得出K因子。 美国国家标准局The National Institute of Standards and Technology(NIST)提供TEM标准样品。 目前国内可以提供SEM标准样品,但还没有TEM标样。
DT =(1-Rout/Rin)X100% 这里Rout和Rin分别是处理器输出和输入计数率。 活时间是测量电路能够检测X射线光子的时间。活时间+死时间 就是实际中的采集EDS谱的时间。 设定活时间是为了使EDS谱峰得到有意义的统计计数。 (1)痕量元素的情况下,活时间必须足够长。应大于100秒。 (2)不稳定样品、导电性差的样品,需减小束流和活时间。
学术干货谈谈能量色散X射线谱仪(EDS)的那些事儿
学术干货谈谈能量色散X射线谱仪(EDS)的那些事儿大家对能够进行样品的微区结构与形貌分析的扫描电子显微镜(SEM)和透射电子显微镜(TEM)都不陌生,而与之相关的利用特征X射线具有特征能量这一原理设计的用于成分分析的能量色散X射线谱仪(EDS),因为不常用,所以可能就没那么熟悉了。
而今天,小编就给大家讲讲,EDS的那些事儿!一、EDS所用信号:高速运动的电子束轰击样品表面,电子与元素的原子核及外层电子发生单次或多次弹性与非弹性碰撞,有一些电子被反射出样品的表面,其余的渗入样品中,逐渐失去其动能,最后被阻止,并被样品吸收。
在此过程中有99%以上的入射电子能量转变成热能,只有约1%的入射电子能量从样品中激发出各种信号。
其中,特征X射线是高能电子激发原子的内层电子,使原子处于不稳定态,从而外层电子填补内层空位使原子趋于稳定的状态,在跃迁的过程中,直接释放出具有特征能量和波长的一种电磁辐射,即特征X射线。
图1:高能电子轰击样品表面所能产生的各种信号二、能量色散X射线谱仪(EDS)的结构与工作原理不同元素发射出来的特征X射线能量是不相同的,利用特征X射线能量不同而进行的元素分析称为能量色散法。
所用谱仪称为能量色散X射线谱仪(EDS),简称能谱仪。
图2:能谱仪结构及工作原理X射线能谱仪的主要构成单元是Si(Li)半导体检测器,即锂漂移硅半导体检测器和多道脉冲分析器。
能量为数千电子伏特的入射电子束照射到样品上,激发出特征X射线,通过Be窗直接照射到Si(Li)半导体检测器上,使Si原子电离并产生大量电子-空穴对,其数量与X射线能量成正比。
这是因为:•产生一个空穴对的最低平均能量为ε•则由一个X射线光子造成的电子空穴对的数目为:N=ΔE/ε•入射X射线光子的能量越高,N就越大。
•不同元素发射不同能量的X射线,不同能量的X射线将产生不同的电子空穴对数。
例如:Fe的Kα辐射可产生1685个电子空穴对,而Cu为2110个。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(4)L壳层分为三个亚壳层,最外面的亚层的电子跃迁到K层产生 的特征X射线称为Kα1,次外层产生的特征X射线称为Kα2。依此类推。 虽然EDS可以记录所以特征X射线,但一般只表征K,L,M和α,β。
X射线荧光产额ωk
原子内壳层电子电离过程中产生特征X射线的几率。
特征X射线的荧光产额ωk与俄歇电子产额ak的关系为 Ωk + ak =1 例如 Al(Z=13),其ωk≈0.04;ak≈0.96 C(Z=6), 其ωk≈0.001;ak≈0.999
韧致辐射(连续X光)
入射电子穿越原子外壳层与原子核产生非弹性碰撞, 与原子库伦场的交互作用使电子损失一定能量,损失 的能量会以X光的形式释放出来。由于损失的能量取 决于非弹性碰撞的大小,所以X光的能量是连续的, 甚至可以高达入射电子的能量。这种连续的X光称为 韧致辐射。 韧致辐射的强度取决于样品中平均的原子序数Z。在 EDS谱中形成连续的背底,而特征X射线形成一个个不 连续的窄峰,是EDS成份分析的基础。
K AB
K AC K BC
K因子理论发展:质量厚度因子,但需准确指导束流强度
M. Watanabe and D.B. Williams, J. Micro., Vol. 221, 2006, pp 89.
(二)获取EDS谱
(1)首先,应尽量利用Ka谱线,因为L和M谱线中重叠 谱峰太多,除非是Ka谱线对Si(Li)探头来说能量太高,比 如原子序数比较高的元素。 (2)在样品不发生漂移或者污染的情况下,获取尽可 能高的特征X射线计数率。
束发散b
由于弹性散射使得电子束在样品中传播时产生展宽效应
E0:电子束能量;t: 样品厚度;ρ:样品密度;
加速电压越高,薄膜材料 的空间分辨率就越高。
加速电压越高,体材料的 空间分辨率就越差。
空间分辨率R
如果入射电子束强度 分布符合高斯分布
R
d Rmax 2
d Rmax R 2
其中
1 2 2
X射线计数N越大,则相对误差越小。
本节重点
(1)空间分辨率和最小探测质量的概念 (2)判定某种痕量元素是否存在的判据
§4.2.4 X光信号的探测过程
在样品中不同元素发出的不同能量E的特征X射线光 子进入Si(Li)探测器以后,就在Si(Li)晶体内产生电子- 空穴对。在低温条件下(液氮温度-196oC冷却条件下, Si(Li)晶体温度约为-170oC到-180oC)产生一个电子- 空穴对的能量ε为3.8eV。能量为E的X射线光子进入 Si(Li)晶体激发的电子-空穴对数量为N=E/ε。 例如:Mn Ka的能量为 5.895keV,形成的电子-空穴 对数为1550个。
这里KAB称为Cliff-Lorimer K因子,K因子不是常数。 K因子与加速电压、样品成份、厚度、EDS探测器的效率 等因素有密切关系。
(1)在两种元素的情况下,CA+CB =100% (2)在多种元素的情况下,
CC IC K CB CB IB
并且CA+CB+CC =100% (3)K因子对应于不同的元素对,如AB,BC,AC等
第四章 能量色散X光谱EDS
目录
§4.1 绪论 §4.2 EDS原理与分析
§4.2.1 §4.2.2 §4.2.3 §4.2.4 特征X射线的产生 EDS分析原理 空间分辨率和最小探测质量 X光信号的探测过程
§4.1 绪论
EDS 70年代初问世以来,发展速度很快:
(1)分辨率已达到130eV左右,理论分辨率约100eV
(3)使样品的倾转为零,并且样品的刃边对着EDS探头。 (4)如果样品处于动力学衍射的双束条件,应稍微倾转 样品,使得样品偏离双束条件。
(三)扣除背底
所谓背底是指EDS谱中特征峰下面由韧致辐射造成的 连续强度分布。背底的强度随X射线能量的增加而减小。 双窗口法
在特征X射线峰的两侧取等宽的两个 窗口,两窗口积分值的平均值作为背 底强度。 适用与能量大于2keV的特征X射线峰, 并且样品很薄韧致辐射不被吸收的情况
吸收校正
吸收校正和荧光校正
当样品比较厚,或者特征X射线的能量小于1keV(轻 元素)的情况下,从样品射出的X射线要受到样品本身 的吸收,X射线的吸收效应就应该加以考虑, 做吸收校正。
I I 0 exp( ut )
这里u:吸收系数; ρ:样品密度; t:长度
吸收校正是最重要的校正
荧光校正
荧光校正,某些高能的X射线如果其能量大于其他元素 的临界电离能,就可使这些元素电离,产生二次X射线, 即荧光X射线,所以需要做荧光校正。 X射线吸收和荧光X射线是 密切相联的,X射线的吸收 往往就意味着荧光X射线的 产生。
ห้องสมุดไป่ตู้
(2)以前Be窗口元素分析范围为11Na-92U,现在一 般都用有机膜超薄窗口,分析元素可从5B-92U。
(3)较完善的元素定性、定量分析等软件,定量准 确度也有很大提高,中等原子序数元素基本无重叠峰, 定量准确度已接近于波谱仪。
收集角Ω (Collection angle) 反映了探测器的收集效率
(2)采集特征X光信号的时候,注意以下几点: (a)一定要取出物镜光栏; (b)一般样品处于倾转为零的状态; (c)EDS模式下合轴,仔细调节聚光镜光栏位臵; (d)尽量利用样品薄区获取X光信号; (d)用Be样品台。
本节重点
采集X光信号应注意的事项
§4.2 EDS原理与分析
§4.2.1 特征X射线的产生
探测器输出的电压脉冲高度,由电子-空穴对的数量 N决定,形成EDS谱的横坐标-能量。 前臵放大器放大的信号,进入多道脉冲高度分析器, 把不同能量的X射线光子区分开来,在EDS谱上形成 纵坐标-脉冲数。
X光信号的处理过程
EDS元素分析范围
EDS分析的元素范围一般是从硼B(Z=5)到铀U(Z=92)。 氢和氦只有K层电子,不能产生X射线。锂虽然能产生X 射线,但其波长太长,无法检测。铍(Be)的X射线产额 太低,EDS探头窗口对Be的吸收严重,另外Be有毒,所 以Be含量很难检测到。 Oxford牛津的INCA EDS会自动选择线系。 (1)元素的原子序数Z<32(Ge)时,选择K线系Ka,因为 Ka的强度大于Kβ; (2)元素的原子序数32≤Z≤72(Hf)时,选用L线系; (3)当Z≥72(Hf)时,选用M线系。 (4)为避免样品中各元素之间的干扰(峰重叠),也 可选用其他线系。
DT =(1-Rout/Rin)X100% 这里Rout和Rin分别是处理器输出和输入计数率。 活时间是测量电路能够检测X射线光子的时间。活时间+死时间 就是实际中的采集EDS谱的时间。 设定活时间是为了使EDS谱峰得到有意义的统计计数。 (1)痕量元素的情况下,活时间必须足够长。应大于100秒。 (2)不稳定样品、导电性差的样品,需减小束流和活时间。
意味着激发1000个C原子才能产生一个特征X光信号。
X射线荧光产额ωk与原子序数的关系
随原子序数Z的增加,特征X射线的荧光产额ωk也增加。
(1)临界电离能量EC(Critical Ionization Energy) 激发原子内壳层电子使原子电离的最小能量。 EC随原子序数增减而增加。 (2)电离截面σ(Ionization Cross Section) 单位面积内的电离数目。电离截面是电离事件发生几率 的数学描述。 在透射电镜中,主要考虑过压比Overvoltage U,也就是 电子束能量和临界电离能Ec的比值。一般Ec<20keV,而 入射电子能量≥100keV, 所以U≥5,电离截面一般是个 常数。如果入射电子能量接近或者稍大于临界电离能, 则电离几率很小。
(2)采集速率(Acquisition Rate)
处理系统每秒可处理的X射线光子数,单位是cps (Counts per second),通常指脉冲处理器的输出 计数率。如果计数率太低,则需要较长的采集时 间,样品漂移和污染就有可能造成问题。如果计 数率太高,则有可能出现和峰。
EDS谱采集过程中的重要参数
(1)死时间(Deadtime)和活时间(Live time)
计数测量系统处理一个脉冲信号后,恢复到能处理下一个脉冲 信号所需要的时间,称为死时间,简称DT。 通常EDS探测器接受大量的X射线光子,但系统脉冲处理器在某 段时间内只能处理一个先期到达的光子,通道处于关闭状态, 拒绝下一个到达的脉冲进入,并将其排斥掉,反而造成输出计 数率的下降。死时间常常用所占总时间的百分数来表示。
利用标准样品确定K因子
所谓标准样品,就是成份已知,且样品厚度也知道的 TEM样品。 利用标准样品,测得IA和IB的值,由于成份CA和CB已知 则利用Cilff-Lorimer公式就可得出K因子。 美国国家标准局The National Institute of Standards and Technology(NIST)提供TEM标准样品。 目前国内可以提供SEM标准样品,但还没有TEM标样。
Rmax (b d )
2
(二)最小探测质量 Minimum Detectability
最小探测质量是在可分析的体积内可探测到的原子数 一般认为某种元素特征X射线的强度N等于背底标准 偏差的的3倍时,即N=3 X B 时,改元素肯定存在, 其臵信度为99.7%。 这里 B 是背底的统计涨落。
本节重点
(1)特征X射线和韧致辐射 (2)La 代表什么谱线?
§4.2.2 EDS分析原理
(一)Cliff-Lorimer因子,简称K因子
假设样品中含有两种元素A和B,EDS谱中元素A和B的强度 分别为IA和IB,在不考虑吸收和荧光校正的情况下,则元 素A和B的重量百分比CA和CB的关系为
CA IA K AB CB IB
(四)K因子的确定
K因子不是常数,它随电镜、EDS探测器(包括窗口、 固体收集角、取出角等)、样品厚度和成份、获取EDS 谱的收集条件、EDS谱的分析和处理条件等有密切关系。 只有在以上条件都相同的条件下K因子才是常数。如果 条件改变,则K因子也随之改变。