质粒载体的构建
质粒构建实验步骤1

质粒构建实验步骤实验一1.将合成的引物溶解(10uM),分别扩增P1P2,P3P4,2.PCR扩增条件如下:95 ℃ 10 min95 ℃ 30 s50℃ 30 s 10cycle72℃ 30 s72℃ 5min3.反应体系为:20 ul (p1p2和p3p4各两管)dNTP 2 ul10xBuffer 2ulTaq 0.4 ulPrimer P1 (P3) 1.0 ulPrimer P2 (P4) 1.0 ulH2O 13.6 ul4.将产物电泳,检测是否为目标片断(分别为A1B、A2B),证实为目的片断以后,进行后续扩增5.将扩增出的AB和CD片断等摩尔加入作为模板进行第二次PCR,并且在第二次PCR 时加入引物扩增25个循环,PCR扩增条件如下:95 ℃ 15 min95 ℃ 30 s60 ℃ 30 s 25cycle72℃ 40 s72℃ 5min6.反应体系为:100uldNTP 10 ul10xBuffer 10 ulTaq 1ulProduct P1P2 2 ulProduct P3P4 2 ulPrimer F 1 ulPrimer R 1 ulH2O 73ul实验二连接载体DNA外源DNA片段10×T4 DNA ligase bufferT4 DNA ligase 0.5μl16℃保温8-24小时。
做二组对照反应,其中对照组一只有质粒载体无外源DNA;对照组二只有外源DNA片段没有质粒载体。
实验三10.转化①取100ul感受态细胞置于冰上融化,将50ul感受态细胞加入至10ul连接产物中,冰上30min。
②42℃放置45~60s,冰上放置2~3min。
③加入37℃预温好的200ul LB液体培养基(不含Amp)④37℃振荡培养1h(160~220 rpm)11.铺板铺板之前要先将AMP抗性的LB固体培养基先预温到常温,然后取100 ul菌液涂板,37℃培养过夜(16~24h)12.不带有质粒DNA的细胞,由于无Amp抗性,不能在含有Amp的筛选培养基上成活。
质粒的构建

② -半乳糖苷酶Xgal显色反应: -半乳糖苷酶能把无色的化合物 Xgal分解成半乳糖和一个深蓝色的 物质5-溴-4-氯靛蓝。 -半乳糖苷酶 Xgal 半乳糖
5-溴-4-氯靛蓝
③ lacZ的肽互补 1)-肽( lacZ’ ): -半乳糖苷酶N端的一段氨基酸片断 (11-41氨基酸)。 N端的11-41aa N端的11-41aa N端的11-41aa N端的11-41aa C端大部分 C端大部分 C端大部分 C端大部分
(2)长度
4363bp
(3)选择标记 氨苄青霉素和四环素抗性。
(4)克隆位点
24个克隆位点。
其中9个会导致Tetr基因失活(如 BamH I、Hind Ⅲ、Sal I); 3个会导致Ampr基因失活(Sca I、 PvuI、Pst I)。
(5)pBR322的筛选 ①双抗菌素抗性选择标记
在EcoRⅠ和HindⅠ酶剪切位点之间插入外源基因
(2)长度
(3)克隆位点
约2.7kb
10个连续的单一限制酶切位 点,位于lacZ’基因的5’端。
ห้องสมุดไป่ตู้
pUC18/19
选择标记 Ampicillin 抗性和 lacZ的肽互补(蓝白 斑)相结合。 筛选方法
选用带有含有ampicillin和X-gal的培养基 受体菌lacZ突变(lacZ∆M15)
受体菌lacZ突变(lacZ∆M15) 受体菌基因组的-半乳糖苷酶基因的 氨基端有缺失(缺失肽),不能形成 4聚体的活性酶,不能分解Xgal 受体菌株:JM系列、TG1、TG2、 XL1-blue、XS127、XS101、KK2186、 MV1184、DH5a
质粒载体生物学特征
分类 按复制类型可分为:松弛型 严紧性 按转移性可分为转移型和非转移型
质粒构建课件

Digestion & ligation
PCR product or vector Enzyme 1 Enzyme 2 10XBuffer 100XBSA(?) ddH2O
37°C 2~3 h
PCR product Vector T4 DNA ligase 10Xligase buffer ddH2O
pCS2(GFP)
……
/vectordb/
Gene cloning
Target gene: hBex2 Vector selection Primer design Amplification, digestion & ligation Transformation & Identification
AAT TCG AAG
CGG TG
EcoR I/BamHI digestion
BamH I GAT CCA CCG
GFP
G AAT TCG AAG
insert
CTT CG A ATT C
GCG GAT CCA CCG
CGG TGG ATC CGC
GFP
Gene cloning
Target gene: hBex2 Vector selection Primer design Amplification, digestion & ligation Transformation & Identification
质粒构建
孟庆明 2019-04-09
1
1
主要内容
质粒的概念及特点 质粒构建的基本步骤和原理
一、质粒的概念
载体(vector;carrier;vehicle) 可以插入核酸片段、能携带外源核酸进入宿主细胞,并在其中进行 独立和稳定的自我复制的核酸分子。基因工程中广泛应用的载体多 来自人工改造的细菌质粒、噬菌体或病毒核酸等。多数载体是DNA 分子,但某些RNA分子也能用做载体。
重组表达质粒的构建——原核表达载体选择
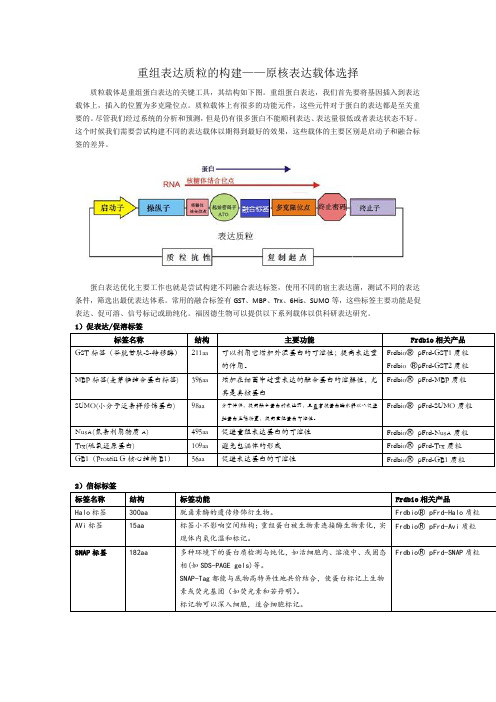
重组表达质粒的构建——原核表达载体选择质粒载体是重组蛋白表达的关键工具,其结构如下图。
重组蛋白表达,我们首先要将基因插入到表达载体上,插入的位置为多克隆位点。
质粒载体上有很多的功能元件,这些元件对于蛋白的表达都是至关重要的。
尽管我们经过系统的分析和预测,但是仍有很多蛋白不能顺利表达、表达量很低或者表达状态不好。
这个时候我们需要尝试构建不同的表达载体以期得到最好的效果,这些载体的主要区别是启动子和融合标签的差异。
蛋白表达优化主要工作也就是尝试构建不同融合表达标签,使用不同的宿主表达菌,测试不同的表达条件,筛选出最优表达体系。
常用的融合标签有GST、MBP、Trx、6His、SUMO等,这些标签主要功能是促表达、促可溶、信号标记或助纯化。
福因德生物可以提供以下系列载体以供科研表达研究。
1)促表达/促溶标签2)信标标签3)纯化标签我们选择表达载体的时候不但要考虑蛋白怎么表达成功,更要考虑蛋白怎么纯化出来,纯化的问题主要是考虑纯化标签和酶切位点的选择,下表我们列举了常见的纯化标签和酶切位点。
4)酶切位点以上为原核表达常用的标签和酶切位点,其性质也都作了简要的介绍,各专业网站或专业书籍已对此做详尽解释,科研工作者可根据具体实验设计方案,组合设计以上标签和酶切位点的使用。
特别值得注意的是,选用和设计蛋白酶切位点的时候首要考虑的是序列内部有没有蛋白酶位点,同时要考虑酶切的效率和蛋白酶试剂成本。
一般商业化载体,在标签蛋白与载体多克隆位点之间都设计有酶切位点。
标签可设计在N-端也可在C-端,设计在N-端的优势是,可通过标签高效翻译起始位点带动插入蛋白的表达,可溶性标签的高效表达更可促进蛋白的可溶性表达;同时,大部分的蛋白内切酶的切割位点在C-端,所以标签设计在N-端可将标签切割完全。
在设计标签序列与酶切位点的时候还要考虑N-端稳定性原则,也就是所谓宿主细胞的N-端规则(N-end rule),这个要避免;同时,还应该检查是否引入了可与别的蛋白相互作用的序列或者蛋白酶切位点。
载体构建介绍

5.常见问题
选择标记类型和选择 1、选择性标记类型 药物抗性(如Kan,Amp等) 营养依赖性标记(如SC-Leu 等) 2、如何选择 根据所选载体上所带的标记
5.常见问题
Gateway系统引物设计-需要对读码框
这是因为中间载体和终载体上某些编码氨基酸或者抗性 基因同目的片段共用一个起始密码子。
载体构建
戎浩 2015.12
C
ONTENTS
目录
1 2 3 4
载体简介 载体构建
注意事项 常见问题
1.载体简介
目的基因的克隆与鉴定
ห้องสมุดไป่ตู้
生理检测 纯化
载体构建
大肠转化,质粒 提取与鉴定
移栽
分子检测
继代繁殖 农杆菌的转化与活化
外植体制备
筛选
浸
染
共培养
1.1载体
载体(vector) ,能将外源DNA或基因片段携带入 宿主细胞内的一个具有自我复制能力的DNA分子。
退火温度不合适,设置梯度,选择最适退火温度
2、条带不单一
引物不特异,适当增长引物序列长度;
适当提高退火温度
5.常见问题
载体酶切的问题 1、酶切质粒浓度和纯度要好 2、酶切温度和时间
如果两个酶的最适温度不同,建议单酶切,回收后在
用另一个酶切,时间最好过夜切。 3、没有切开 可能是酶失活,建议酶切时增加阳性对照,确定酶是 否好用
5.常见问题
克隆基因的酶切位点及引物问题 1、单酶切 单酶切后进行连接,质粒自连、目的片段自连、目的
片段之间连接、目的片段和载体各种错误连接、目的片段
反向连接等等,尽量不选单酶切 2、保护碱基数目的问题。 在设计PCR引物时,引入酶切位点后,常常要加入保 护碱基,这会使得酶切效率大大提高。
质粒构建

PCR(多聚酶链式反应)一、实验原理PCR用于扩增位于两端已知序列之间的DNA区段,即通过引物延伸而进行的重复双向DNA合成。
基本原理及过程如下:PCR循环过程中有三种不同的事件发生:(1)模板变性;(2)引物退火;(3)热稳定DNA聚合酶进行DNA合成。
1、变性:加热使模板DNA在高温下(94-95℃)变性,双链间的氢键断裂而形成两条单链,即变性阶段。
2、退火:在体系温度降至37-65℃,模板DNA与引物按碱基配对原则互补结合,使引物与模板链3’端结合,形成部分双链DNA,即退火阶段。
3、延伸:体系反应温度升至中温72℃,耐热DNA聚合酶以单链DNA为模板,在引物的引导下,利用反应混合物中的4种脱氧核苷三磷酸(dNTP),按5’到3’方向复制出互补DNA,即引物的延伸阶段。
上述3步为一个循环,即高温变性、低温退火、中温延伸3个阶段。
从理论上讲,每经过一个循环,样本中的DNA量应该增加一倍,新形成的链又可成为新一轮循环的模板,经过25~30个循环后DNA可扩增106~109倍。
典型的PCR反应体系由如下组分组成:DNA模板、反应缓冲液、dNTP、MgCl2、两条合成的DNA引物、耐热DNA Taq聚合酶。
二、按下列组份配制PCR 反应液TaKaRa LA Taq(5 U/μl)0.5 μl10×LA PCR Buffer II(Mg2+ Plus) 5 μldNTP Mixture(各2.5 mM)8 μl模板DNA(λDNA)* 2.5 ng引物1(10 μM) 2 μl引物2(10 μM) 2 μl灭菌蒸馏水up to 50 μl*【50 μl PCR反应体系中模板DNA 推荐使用量】人基因组DNA 0.1 μg~1 μg大肠杆菌基因组DNA 10 ng~100 ngλDNA 0.5 ng~5 ng质粒DNA 0.1 ng~10 ng三、PCR 反应条件。
以λDNA 为模板,扩增 1 kbp、35 kbp 的DNA片段的PCR 反应条件如下:【1 kbp】95℃ 5 min.95℃30 sec.55℃30 sec. 35 Cycles72℃ 1 min.72℃10 min.【35 kbp】94℃ 5 min.98℃10 sec.68℃15 min. 35 Cycles72℃10 min.常用循环:95℃ 5 min.95℃45 sec.55℃45 sec. 35 Cycles72℃ 2 min.72℃10 min.四、可能出现的问题与解决方案:1、假阴性,不出现扩增条带PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量,④PCR循环条件。
质粒载体的构建及其在基因工程中的应用研究

质粒载体的构建及其在基因工程中的应用研究随着基因工程技术的不断发展,质粒载体成为了实现基因转化的重要手段之一。
质粒载体构建是基因工程研究的重要环节,其成功与否直接关系到后续研究的开展和成果的取得。
本文将介绍质粒载体的构建原理、构建方法以及其在基因工程中的应用研究。
一、质粒载体的构建原理质粒载体是人工合成的单链圆形DNA分子,作为存储DNA序列的一种手段,其基本结构特征主要包括起始端、终止端、多个酶切位点、标记位点等。
考虑到质粒载体在基因工程研究中的应用,构建原理主要包括以下几点:1. 合成目的基因的DNA序列,包括启动子、编码区、终止子等。
2. 找到合适的质粒载体,通过酶切识别并将目的基因DNA序列与质粒载体进行连接。
3. 对连接后的质粒载体进行转化,将其转移到细胞中,获得外显表达的蛋白。
质粒载体的构建原理相对简单,但质粒载体在实际应用中的构建过程则需要复杂的技术手段和严密的实验操作。
二、质粒载体构建方法1. 基于PCR扩增法PCR扩增法是目前基因工程研究中最常用的方法之一。
选择需要进行扩增的目的基因DNA序列,使用酶切产生目的碎片,通过PCR扩增后,利用限制性内切酶等酶切方法进行连接。
2. 基于化学合成法在基于化学合成法中,研究者可以通过化学合成方式来合成目的基因DNA序列,在此基础上通过PCR扩增和限制性内切酶等酶切方法进行连接。
3. 基于网站选择法基于网站选择法是现在比较流行的方法之一,具有操作简单、成本低等优点。
研究者可以在网站上选择目的基因序列,并结合实验室已有的质粒载体库,在网站上进行设计、合成、定制和质粒表达等步骤。
三、质粒载体在基因工程中的应用研究质粒载体在基因工程中的应用研究十分广泛,可以用于植物基因转化、动物基因转化、疫苗研发、DNA疫苗的制备、表达蛋白的研究等方面。
1. 植物基因转化通过基因转化技术向植物中加入外源基因,可以使植物表现出新的性状或特征。
在实践中,研究者会将需要转入的序列合成后,利用限制性内切酶或其他酶切方法将其插入到质粒载体中,再利用农杆菌等工具将质粒载体导入植物细胞中,从而实现植物基因转化。
质粒载体的概况及构建 ppt课件

ppt课件
12
④ 用连接产物转化大肠杆菌感受态细胞时转化细菌 有两个基本方法:
一是化学法,即用CaCl₂处理并加热激以促进DNA进入 菌体;
二是电激法,即施加短脉冲的电荷以促进DNA吸收。
ppt课件
13
⑤ 筛选时会有三种结果:
一是要插入的序列自连,结果没有菌落形成;
二是切开的质粒重连,结果表现为蓝色菌落;
三是要插入的序列与质粒相连产生白色的抗氨苄青霉素菌 落。
pptቤተ መጻሕፍቲ ባይዱ件
14
THANKS
ppt课件
15
(2)不相容性 同一复制系统的不同质粒在同一细菌中不能相容 不同复制系统的质粒在同一细菌中可共存
ppt课件
5
(3)可扩增性
• 质粒能自主地进行复制,这样它才能随着细菌细胞的 分裂而稳定地遗传。
• 质粒就其复制方式而言分为两类:松弛型复制及严谨 型复制。
• 松弛型复制:拷贝数高,一个细胞中可达上千个拷贝;
★ 质粒不是细菌生长所必须的 ★ 可赋予细菌抵御外界因素不利影响的能力 ★ 分子量在1~200kb之间
ppt课件
3
质粒(plasmid)
特点:
能在宿主细胞内独立复制;带有某些遗传信息,会赋予
宿主细胞一些遗传性状。 ppt课件
4
(二)质粒的基本特性
(1) 自主复制性 质粒DNA携带有自己的复制起始区(ori) 控制质粒拷贝数的基因 能独立于宿主细胞的染色体DNA而自主复制
ppt课件
8
质粒的制备
实验室一般使用两种方法制备质粒: (一)碱裂解法 (二)沸水浴法
ppt课件
9
(1)碱裂解法
原理:利用溶菌酶在NaOH与SDS的混合溶液中破坏细菌 外壁,去除染色体DNA及变性蛋白质,离心,苯酚—氯仿 溶液处理,乙醇或异丙醇沉淀水相质粒。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
四、连接 很多公司都有快速连接试剂盒和普通试剂盒。一般都是 16 度数个小时或者过夜,或者 4 度 过夜,不一而同。曾在园子里看到一个高人的文章,认为是时间越长越好,甚至建议 4 度放 几天,我没有试过。16 度可以用 PCR 仪创造,或者找一个泡沫盒,加上冰,水搞到 15 度 左右,放入 4 度冰箱即可。我用的 TAKARA 的快速连接试剂盒,4 度过夜。 菜鸟体贴提示: 1、 通过电泳,粗略判定质粒和 DNA 的浓度比例。一般加入连接液的 DNA:质粒=9:1 2、 放入质粒和 DNA 的总量要适当,不可超过说明书,宁少不要多。
质粒载体的构建-菜鸟入门手册
dongkey 制作
本人刚刚完成质粒载体的构建,总结了一下,以方便要做这方面的菜鸟同学借鉴一下,希望 老鸟同学批评指教。 一、确定插入的基因片段。 首先要确定自己要插入载体中的基因片段,比如要做蛋白表达和功能,可以选择 CDS 区进 行插入。然后自然是如何得到这段基因片段的问题了。最常见的是 PCR 解决了。那么就涉 及到引物的设计。插入 CDS 基因片段的例子:找到 CDS,根据 CDS 设计全长引物,然后 加上内切酶的碱基片段(这要根据自己手头上有的质粒的内切酶位点来决定了),注意,前 面要加上保护碱基!然后进行 PCR。 PCR 后要跑电泳确定目的基因的长度是正确的。 用乙醇沉淀法纯化 PCR 产物(具体见分子克隆一书) 菜鸟体贴提示:要在质粒上找到两个最好不是连续在一起的内切酶,然后分别添加在两条引 物的 5‘端,当然内切酶的温度最好是一致的,而且可以能够同时切开的(双酶切)。这可 以在试剂公司的限制性内切酶的列表上找到有没有共同的 BUFFER 及其双酶切时的活性如 何(当然是越大越好了)。内切酶的公司一般可以找 NEB, TAKARA, TOYOBO 等公司,本 人用的是 TAKARA。 二、酶切 将质粒和基因片段分别进行酶切。最省事的是同时双酶切了。一般酶切温度都是 37 度。 菜鸟体贴提示: 1、 确定质粒和基因片段的量!!(宁少不多),根据说明书加样,一般是 DNA+BUFFER+
加内切酶。注意 BUFFER 的比例,有的是 1×,有的是 1.5×。 2、 确定内切的时间。这是比较讨厌的环节。切的时间不能短,也不能太长。最好是做一个
梯度。注意,说明书的内切时间不一定就是标准的!!(本人经验是可以稍微切的时间长 一点,也不用短了) 3、 确定酶切成功了。 对于质粒,可以将空质粒和切开的质粒同时电泳跑,对比一下就知道了 对于目的基因,很难,因为切开的是几个碱基而已,不过一般如果质粒切开,目的基因 应该也会被切开的。 酶切完成后,有两种方法回收 1、 电泳跑胶回收 2、 不跑胶,直接用纯化试剂盒回收。 根据 N 多种理论,很多人建议用第一种方法较好。理由这里就不罗列了。
五、转化 按照分子克隆的步骤来。 如果是买公司的感受态,根据公司的说明书。 菜鸟提示: 1、复苏的时间一定要掌握好,还有是装感受态的管子要薄,以有利于温度的传导。一般用 恒温水浴。 2、涂抹在培养皿上的时候,可以做一个梯度,200ul,300ul,400ul,一定要晾干。
六、确定测序的克隆。 一般在培养皿上挑 10-30 个左右的单克隆。加入培养液(含抗生素)过夜培养,后用 2ul 的培养液,加上步骤一的引物,进行 PCR,电泳,看有没有目的基因的条带。 然后将有条带的培养液提取质粒,双酶切,电泳看看有没有目的基因的条带。如果有的话, 可以送去测序了。 菜鸟提示: 1、 可以绕过 PCR 直接提取质粒酶切(如果挑的克隆太多的话,建议还是先 PCR 以缩小范
围)。 2、 如果目的基因较小的话(如 100-300bp),那么酶切后电泳加样要尽量多一点,因为切
下来的目的基因的亮度对比质粒的亮度是 100-330/数千 bp 分之一。
七、测序结果的分析 可以使用多种软件。这里不罗列了。 菜鸟方法:一般公司发回来的有记事本或者 WORD 的排列好测序结果的序列。将它们复制 到一个新的 WORD 文档中,调整好格式,如全部大写,没有空格,没有回车符号等。然后, 将目的基因全长 COPY,使用查找功能,看是否全符合。