NCBI在线BLAST使用方法与结果详解
图解blast验证引物教程

图解blast验证引物教程1、进入网页:/BLAST/2、点击Search for short, nearly exact matches3、在search栏中输入引物系列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物B、输入上游引物回车输入下游引物4、在options for advanced blasting中:select from 栏通过菜单选择Homo sapiens【ORGN】Expect后面的数字改为105、在format中:select from 栏通过菜单选择Homo sapiens【ORGN】Expect后面的数字填上0 106、点击网页中最下面的“BLAST!”7、出现新的网页,点击Format!果。
(1)图形格式:图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:从左到右分别为:A、数据库系列的身份证:点击之后可以获得该序列的信息B、系列的简单描述C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
按照得分的高低由大到小排列。
得分的计算公式=匹配的碱基×2+0.1。
举例:如果有20个碱基匹配,则其得分为40.1。
Blast分析报告

Blast分析报告引言Blast(Basic Local Alignment Search Tool)是一种常用的生物信息学工具,用于比对和比较生物序列。
本报告旨在分析和解释Blast结果,帮助读者理解序列的相似性和演化关系。
方法为了进行Blast分析,首先需要准备两个序列:查询序列和参考序列。
查询序列是我们要研究的序列,而参考序列是已知的序列。
Blast会将查询序列与参考序列进行比对,并计算序列之间的相似性。
在本次分析中,我们使用了NCBI(National Center for Biotechnology Information)提供的在线Blast工具。
具体的分析步骤如下:1.登录NCBI网站并进入Blast页面。
2.将查询序列输入到指定的文本框中。
3.选择参考序列数据库。
4.点击“运行Blast”按钮,等待分析结果。
结果经过Blast分析,我们获得了以下结果:1.序列相似性分析:Blast会将查询序列与参考序列进行比对,并计算序列之间的相似性。
结果以百分比的形式表示相似度。
较高的相似度表明序列之间有较高的共同点。
2.演化关系分析:Blast还可以帮助我们了解序列之间的演化关系。
通过比较序列中的保守区域和变异区域,我们可以推断序列的起源和演化路径。
讨论根据Blast分析结果,我们可以得出以下结论:1.查询序列与参考序列的相似性较高。
根据相似性百分比可以判断两个序列之间的关系,例如亲缘关系或功能相似性。
2.查询序列可能与参考序列在演化上存在一定的共同点。
通过比较序列中的保守区域和变异区域,我们可以推断序列的起源和演化路径。
3.查询序列与参考序列之间的差异可能与物种间的差异相关。
通过进一步的分析,可以探究这些差异对生物体功能的影响。
结论本次Blast分析报告旨在帮助读者理解序列的相似性和演化关系。
通过Blast工具,我们可以快速准确地比对和比较生物序列。
通过对结果的分析,我们可以推断序列的起源和演化路径,并进一步探究序列间的差异对生物体功能的影响。
在线blast的用法总结
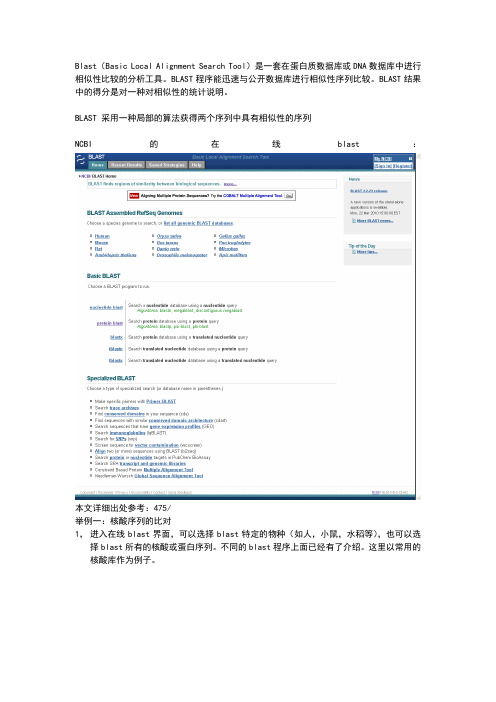
Blast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列NCBI的在线blast:本文详细出处参考:475/举例一:核酸序列的比对1,进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
(补充介绍下:1、BLASTN【 nucleotide blast】是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
2、BLASTP【protein blast】是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
3、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
)2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
NCBI在线BLAST使用方法与结果详解

NCBI正在线BLAST使用要领与截止详解之阳早格格创做BLAST(Basic Local Alignment Search Tool)是一套正在蛋黑量数据库或者DNA数据库中举止相似性比较的分解工具.BLAST步调能赶快与公启数据库举止相似性序列比较.BLAST截止中的得分是对于一种对于相似性的统计证明.BLAST 采与一种局部的算法赢得二个序列中具备相似性的序列.Blast中时常使用的步调介绍:1、BLASTP是蛋黑序列到蛋黑库中的一种查询.库中存留的每条已知序列将逐一天共每条所查序列做一对于一的序列比对于.2、BLASTX是核酸序列到蛋黑库中的一种查询.先将核酸序列翻译成蛋黑序列(一条核酸序列会被翻译成大概的六条蛋黑),再对于每一条做一对于一的蛋黑序列比对于.3、BLASTN是核酸序列到核酸库中的一种查询.库中存留的每条已知序列皆将共所查序列做一对于一天核酸序列比对于.4、TBLASTN是蛋黑序列到核酸库中的一种查询.与BLASTX差异,它是将库中的核酸序列翻译成蛋黑序列,再共所查序列做蛋黑与蛋黑的比对于.5、TBLASTX是核酸序列到核酸库中的一种查询.此种查询将库中的核酸序列战所查的核酸序列皆翻译成蛋黑(每条核酸序列会爆收6条大概的蛋黑序列),那样屡屡比对于会爆收36种比对于阵列.底下是简直支配要领1,加进正在线BLAST界里,不妨采用blast特定的物种(如人,小鼠,火稻等),也不妨采用blast所有的核酸或者蛋黑序列.分歧的blast步调上头已经有了介绍.那里以时常使用的核酸库动做例子.2,粘揭fasta要领的序列.采用一个要比对于的数据库.闭于数据库的证明请瞅NCBI正在线blast数据库的简要证明.普遍的话参数默认.3,blast参数的树立.注意隐现的最大的截止数跟E值,E值是比较要害的.筛选的尺度.末尾会证明一下.4,注意一下您输进的序列少度.注意一下比对于的数据库的证明.5,blast截止的图形隐现.出啥佳道的.6,blast截止的形貌天区.注意分值与E值.分值越大越靠前了,E值越小也是那样.7,blast截止的仔细比对于截止.注意比对于到的序列少度.评介一个blast截止的尺度主要有三项,E值(Expect),普遍性(Identities),缺得或者拔出(Gaps).加上少度的话,便有四个尺度了.如图中隐现,比对于到的序列少度为1405,瞅Identities那一值,才匹配到1344bp,而输进的序列少度也是为1344bp(瞅上头的图),便证明比对于到的序列要少一面.由Qurey(起初1)战Sbjct(起初35)的起初位子可知,5'端是是多了一段的.偶尔也要注意3'端的.附:E值(Expect):表示随机匹配的大概性,E值越大,随机匹配的大概性也越大.E值交近整或者为整时,具原上便是实足匹配了.普遍性(Identities):或者相似性.匹配上的碱基数占总序列少的百分数.缺得或者拔出(Gaps):拔出或者缺得.用"—"去表示.。
NCBI网站BLAST使用方法介绍完整版

息学方法
BLAST
宿主菌
细胞转化
几周的时间 蛋白质分离纯化及性质测定
Gene family Or
Protein Family
几分钟的时间
Function annotation
BLAST
Web Access
Text
Wang LS, Gao PJ, cellulase,et al.
? RPS BLAST
– searches a database of PSSMs – tool for conserved domain searches
Basic Local Alignment Search Tool
? Widely used similarity search tool
? Heuristic approach based on
ACATGGACCCT ...
Protein Words
Query : GTQITVEDLFYNIATRRKALKN
WGoTrdQsize = 3 (default)
TQI
Word size can only be 2 or 3
Make a lookup table of words
QIT ITV
Basic Local Alignment Search Tool
?Why use sequence similarity? ?BLAST algorithm ?BLAST statistics ?BLAST output ?Examples
Why Do We Need Sequence Similarity Searching?
11-mer
GTACTGGACAT
WORD SIZE
NCBI—blast和PDB的使用和介绍

可以输入序列号或FASTA序列 接受默认参数,点击开始搜索
腺苷脱氨酶
分子类型:氨基酸 序列长度:352
已经发现潜在的同源性的领域, 请点击图像下面的详细结果
P03958的保守序列 展示特征序列
其他资源的链接说明 目标序列描述部分
ቤተ መጻሕፍቲ ባይዱ
返回
返回
http://www.ncbi.nlm.n·/BLAS T/ 2.根据数据类型,选择合适的程序 3.填写表单信息 4.提交任务 5.查看和分析结果
在此输入P03958
Result counts displayed in gray indicate one or more terms not found
腺嘌呤核苷/AMP脱 酰氨基酶
生物过程
细胞成 分
谢谢!
PDB的介绍与运用
研究生物大 分子的资源
11个结构 6个引证9个配体 2个网页
显示或下载的 形式
综合报告
digital object 2价锌离子能稳定鼠 identifier: 数字对科腺甙脱氨酶的结构
象标识
酶的分类编 片段 号
分类:水解酶
类型:多 肽
化学配合基 成分
外部域注释
聚合物
脱氨酶 分子功能
NCBI-BLAST 和PDB 的使用和介绍
常用的BLAST工具
核库蛋核质酸中白蛋蛋酸数翻的质白白序据译核-质 质蛋列库成酸序序核白翻中蛋译列列苷质译的白成比-酸核序成序质的对-酸列蛋列核序蛋数比白比苷列白据对质对酸质-核库序序序酸翻列列列数译-比蛋比据后对白对的
BLAST的搜索步骤
1.登陆blast主页
NCBI在线BLAST使用方法与结果详解
N C B I在线B L A S T使用方法与结果详解BLAST Basic Local Alignment Search Tool是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具;BLAST程序能迅速与公开数据库进行相似性序列比较;BLAST结果中的得分是对一种对相似性的统计说明;BLAST 采用一种局部的算法获得两个序列中具有相似性的序列;Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询;库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对;2、BLASTX是核酸序列到蛋白库中的一种查询;先将核酸序列翻译成蛋白序列一条核酸序列会被翻译成可能的六条蛋白,再对每一条作一对一的蛋白序列比对;3、BLASTN是核酸序列到核酸库中的一种查询;库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对;4、TBLASTN是蛋白序列到核酸库中的一种查询;与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对;5、TBLASTX是核酸序列到核酸库中的一种查询;此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白每条核酸序列会产生6条可能的蛋白序列,这样每次比对会产生36种比对阵列;NCBI的在线BLAST:下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种如人,小鼠,水稻等,也可以选择blast 所有的核酸或蛋白序列;不同的blast程序上面已经有了介绍;这里以常用的核酸库作为例子;2,粘贴fasta格式的序列;选择一个要比对的数据库;关于数据库的说明请看NCBI在线blast数据库的简要说明;一般的话参数默认;3,blast参数的设置;注意显示的最大的结果数跟E值,E值是比较重要的;筛选的标准;最后会说明一下;4,注意一下你输入的序列长度;注意一下比对的数据库的说明;5,blast结果的图形显示;没啥好说的;6,blast结果的描述区域;注意分值与E值;分值越大越靠前了,E值越小也是这样;7,blast结果的详细比对结果;注意比对到的序列长度;评价一个blast结果的标准主要有三项,E值Expect,一致性Identities,缺失或插入Gaps;加上长度的话,就有四个标准了;如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp看上面的图,就说明比对到的序列要长一点;由Qurey起始1和Sbjct起始35的起始位置可知,5'端是是多了一段的;有时也要注意3'端的;附:E值Expect:表示随机匹配的可能性,E值越大,随机匹配的可能性也越大;E值接近零或为零时,具本上就是完全匹配了;一致性Identities:或相似性;匹配上的碱基数占总序列长的百分数;缺失或插入Gaps:插入或缺失;用"—"来表示;。
NCBI网站BLAST使用方法介绍完整版
Human genome statistics CCCTAACCCCTAACCCTAACCCTAACCCTAACCCTAACCTAACCCTAACCCTAACCCTAACCCTAAC
21
Seq1: 1 W--HERE 5 W HERE
Seq2: 3 WASHERE 9
Local
Seq1: 1 W--HERE 5 W HERE
Seq2: 15 WISHERE 21
The Flavors of BLAST
? Standard BLAST
– traditional “contiguous” word hit – position independent scoring – nucleotide, protein and translations (blastn, blastp,
AACCCTAACCCTAACCCTAACCCCTAACCCTAACCCTAAACCCTAAACCCTAACCCTAACCCTAACC ACCCTAACCCCAACCCCAACCCCAACCCCAACCCCAACCCCAACCCTAACCCCTAACCCTAACCCTA CTACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCCTAACCCCTAACCCTAACCCTAACC ACCCTAACCCTAACCCTAACCCCTAACCCTAACCCTAACCCTAACCCTCGCGGTACCCTCAGCCGGC CCCGCCCGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAGAGTACCACCGAAATCTGTGCAGAG AACGCAGCTCCGCCCTCGCGGTGCTCTCCGGGTCTGTGCTGAGGAGAACGCAACTCCGCCGGCGCAG CAGAGAGGCGCGCCGCGCCGGCGCAGGCGCAGACACATGCTAGCGCGTCGGGGTGGAGGCGTGGCGC CGCAGAGAGGCGCGCCGCGCCGGCGCAGGCGCAGAGACACATGCTACCGCGTCCAGGGGTGGAGGCG CGCAGGCGCAGAGAGGCGCACCGCGCCGGCGCAGGCGCAGAGACACATGCTAGCGCGTCCAGGGGTG GCGTGGCGCAGGCGCAGAGACGCAAGCCTACGGGCGGGGGTTGGGGGGGCGTGTGTTGCAGGAGCAA CGCACGGCGCCGGGCTGGGGCGGGGGGAGGGTGGCGCCGTGCACGCGCAGAAACTCACGTCACGGTG CGGCGCAGAGACGGGTAGAACCTCAGTAATCCGAAAAGCCGGGATCGACCGCCCCTTGCTTGCAGCC CACTACAGGACCCGCTTGCTCACGGTGCTGTGCCAGGGCGCCCCCTGCTGGCGACTAGGGCAACTGC GCTCTCTTGCTTAGAGTGGTGGCCAGCGCCCCCTGCTGGCGCCGGGGCACTGCAGGGCCCTCTTGCT TGTATAGTGGTGGCACGCCGCCTGCTGGCAGCTAGGGACATTGCAGGGTCCTCTTGCTCAAGGTGTA GCAGCACGCCCACCTGCTGGCAGCTGGGGACACTGCCGGGCCCTCTTGCTCCAACAGTACTGGCGGA TAGGGAAACACCCGGAGCATATGCTGTTTGGTCTCAGTAGACTCCTAAATATGGGATTCCTGGGTTT AGTAAAAAATAAATATGTTTAATTTGTGAACTGATTACCATCAGAATTGTACTGTTCTGTATCCCAC CAATGTCTAGGAATGCCTGTTTCTCCACAAAGTGTTTACTTTTGGATTTTTGCCAGTCTAACAGGTG CCCTGGAGATTCTTATTAGTGATTTGGGCTGGGGCCTGGCCATGTGTATTTTTTTAAATTTCCACTG ATTTTGCTGCATGGCCGGTGTTGAGAATGACTGCGCAAATTTGCCGGATTTCCTTTGCTGTTCCTGC TAGTTTAAACGAGATTGCCAGCACCGGGTATCATTCACCATTTTTCTTTTCGTTAACTTGCCGTCAG
科研实验中的生物信息学工具使用教程
科研实验中的生物信息学工具使用教程生物信息学是将数学、统计学和计算机科学应用于生物学研究的交叉学科。
在现代科研中,生物信息学工具已经成为了生物学实验和研究的重要组成部分。
本文将介绍几种常用的生物信息学工具,并提供详细的使用教程。
1. BLAST(Basic Local Alignment Search Tool)BLAST是生物信息学领域中最常见的工具之一,用于在数据库中快速比较DNA或蛋白质序列的相似性。
以下是使用BLAST进行基本比对的步骤:(1)打开NCBI网站,并进入BLAST页面。
(2)选择“nucleotide”或“protein”,取决于你要比对的序列类型。
(3)复制粘贴或上传你要比对的序列。
(4)选择合适的数据库进行搜索,如“nr”(非冗余数据库)。
(5)点击“BLAST”按钮,等待搜索结果。
BLAST会为你提供一个比对报告,其中包含了与你的查询序列相似的序列列表。
2. EMBOSS(European Molecular Biology Open Software Suite)EMBOSS是一个开源的生物信息学软件包,提供了一系列用于序列分析和比对的工具。
以下是使用EMBOSS进行序列分析的步骤:(1)打开EMBOSS软件(可以下载并安装在你的计算机上)。
(2)选择合适的工具,如“water”(Smith-Waterman比对算法)。
(3)输入查询序列和数据库序列。
(4)设置相关参数,如匹配分数和距离惩罚。
(5)点击“Run”按钮,等待分析结果。
EMBOSS将为你提供一个比对报告,并给出一些统计数据,如匹配分数和最佳比对。
3. R/BioconductorR是一种统计软件和编程语言,Bioconductor是R语言的一个生物信息学扩展包,提供了丰富的生物信息学工具和分析方法。
以下是使用R/Bioconductor进行基因表达分析的步骤:(1)打开R软件并加载Bioconductor包。
NCBI在线BLAST使用方法与结果详解
NCBI在线BLAST使用方法与结果详解NCBI在线BLAST(Basic Local Alignment Search Tool)是一种广泛使用的生物信息学工具,用于比对和分析DNA、RNA或蛋白质序列。
它可以对已知和未知序列进行,找到与查询序列相似的序列,并提供有关相似性和功能的信息。
使用NCBI在线BLAST可以分为四个主要步骤:选择BLAST程序,输入查询序列,选择目标数据库,解析和分析结果。
第一步:选择BLAST程序NCBI提供了多种BLAST程序可供选择,包括BLASTN(DNA对DNA的比对)、BLASTP(蛋白质对蛋白质的比对)、BLASTX(DNA对蛋白质的比对)等。
根据实际需求选择相应的BLAST程序。
第二步:输入查询序列在查询序列的文本框中输入待比对的序列。
可以输入单个序列,也可以上传包含多个序列的文件。
如果输入的序列是DNA或RNA序列,需要选择相应的序列类型。
此外,还可以选择是否使用掩码序列或低复杂性筛选来优化比对结果。
第三步:选择目标数据库用户可以选择目标数据库来与查询序列相似的序列。
NCBI提供了多个常用的数据库,如nr(非冗余蛋白质数据库)、nt(核酸数据库)等。
此外,还可以选择特定的物种数据库来限制比对范围。
第四步:解析和分析结果在BLAST运行完成后,会生成一个结果页面,其中包含了比对结果的详细信息。
结果页面包括比对统计信息、序列比对图、E值、分数等。
通过分析这些信息,可以了解查询序列与目标数据库中的序列之间的相似性和可能的功能。
此外,NCBI在线BLAST还提供了一些高级选项,例如使用特定的算法或参数来进行比对、设置比对阈值、选择比对输出格式等。
这些选项可以根据实际需求进行调整。
总结起来,使用NCBI在线BLAST可以通过选择BLAST程序、输入查询序列、选择目标数据库以及解析和分析结果来比对和分析序列。
通过权衡算法和参数选择,在特定数据库中找到与查询序列相似的序列,从而获得有关其相似性和功能的信息。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
N C B I在线B L A S T使用方法与结果详解
集团企业公司编码:(LL3698-KKI1269-TM2483-LUI12689-ITT289-
N C B I在线B L A S T使用方法与结果详解BLAST(BasicLocalAlignmentSearchTool)是一套在蛋白质数据库或DNA 数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:
1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线BLAST:
下面是具体操作方法
1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
注意分值与E值。
分值越大越靠前了,E值越小也是这样。
7,blast结果的详细比对结果。
注意比对到的序列长度。
评价一个blast结果的标准主要有三项,E值(Expect),一致性(Identities),缺失或插入(Gaps)。
加上长度的话,就有四个标准了。
如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序列要长一点。
由Qurey(起始1)和Sbjct(起始35)的起始位置可知,5'端是是多了一段的。
有时也要注意3'端的。
附:
E值(Expect):表示随机匹配的可能性,E值越大,随机匹配的可能性也越大。
E值接近零或为零时,具本上就是完全匹配了。
一致性(Identities):或相似性。
匹配上的碱基数占总序列长的百分数。
缺失或插入(Gaps):插入或缺失。
用"—"来表示。