富马酸二甲酯英文说明书
ISO TS 16186-2012 鞋类材料中DMFU测定翻译版

技术规范ISO TS16186鞋类--在鞋类和鞋类部件中可能存在的临界物质—在鞋类和鞋类部件中富马酸二甲酯(DMFU)的测试方法。
1.范围这个技术规范提供了一个鞋类及鞋类材料、洗护用品和其他样品中富马酸二甲酯(DMFU)的测试方法。
本方法不可用于金属材料,适用范围见ISO/TR 16178:2012,表1.注意:在欧洲,根据98/8/EC指令,DMFU禁止用于杀虫剂产品。
在化学实验室这个物质必须小心使用以避免任何健康问题。
最近,欧盟委员会通过2009/251/EC指令欧盟成员国禁止在消费品中使用生物杀灭剂富马酸二甲酯(DMFU)和含有富马酸二甲酯(DMFU)的产品投入市场。
该指令规定产品中富马酸二甲酯(DMF)含量必须≤0.1mg/kg。
2.引用标准下列引用标准与本标准的应用是密不可分的。
对于注日期的引用标准,所引用的版本才适用。
对于未注日期的引用标准,所引用标准的最新版本(包括任何修订案)适用于本标准。
ISO 4787,Laboratory glassware—V olmetric instruments –Methods for capacity and for use ISO/TR 16178:2012,Footwear—Critical substances potentially present in footwear and footwear components3.方法原理将样品剪成小块,用丙酮在密闭的容器中用已知的温度超声萃取,在这个过程中,根据不同材料,有以下两种萃取方法可供选取:a)方法一,适用于不需要净化和浓缩就可得到单纯色谱图的样品,如纺织品;b)方法二,适用于需要净化和浓缩处理的复杂基质的样品,如皮革等4.试剂4.1常规试剂需要用到的试剂见表1,物质纯度≥99.5%表1—试剂信息表4.2储备溶液和工作溶液4.2.1内标储备液(1g/l)称量10mg的d2-DMFU转移至10ml容量瓶中,用丙酮定容。
服装环保欧洲标准

Banned Azo colorants 禁用AZO着色剂Harmful substances, heavy metals, and ROHS testing, PAHS (pah), more than POHS, SVHC, halogen, plasticizer (phthalic acid salt), formaldehyde, azo, volatile organic compounds (VOC), asbestos, etc 有害物质,重金属,ROHS,PAHS,POHS,SVHC, 卤素,增塑剂(邻苯二甲酸盐),甲醛,偶氮,VOC(挥发性有机物),石棉等Allergenic and / or carcinogenic colorants 致敏性致癌性染料Perfluorooctane sulfonates (PFOS) 全氟辛烷磺酸(是目前最难降解的有机污染物之一,它具有疏水疏油的特性,用途广泛。
PFOS可以通过呼吸和食用被生物体摄取,其大部分与血浆蛋白结合存在于血液中,其余则蓄积在动物的肝脏组织和肌肉组织中。
动物实验表明,每公斤动物体重有2毫克的PFOS含量就可导致死亡。
欧盟规定若浓度或质量等于或超过0.005%的将不得销售;而在成品和半成品中使用PFOS浓度或质量等于或超过0.1%,则成品、半成品及零件也将被列入禁售范围)Phthalates/softeners according to listing in Oeko-Tex® Standard 100 as well as tris(2-chloroethyl) phosphate 根据Oeko-Tex100标准所列的邻苯二甲酸盐(增塑剂)、软化剂以及二氯乙基磷酸酯Chromium VI 六价铬Cadmium – total content 镉总含量Formaldehyde content 甲醛含量Pentachlorophenol 五氯苯酚(杀虫剂)Flame retardant products --In general not allowed;Usage only after agreement of XX 阻燃剂 --通常不允许,只有在XX同意时才可以用Biological active products 生物活性产品Burning behaviour; house / home textiles --Compliance of adequate requirements 燃烧性能,家用纺织品--符合足够的要求Dimethyl Fumarate 富马酸二甲酯(防霉剂)(Hohenstein in-house method; extraction) (海恩斯坦室内检测法,萃取法)polycyclic aromatic hydrocarbons (PAHs) (16 EPAPAH’s 多(稠)环芳香烃(有致癌作用)+ 8 further substances: +8种更多物质Benzo(j)fluoranthene苯并荧蒽, Cyclopenta(c,d)pyrene环戊二烯并芘,Dibenzo(a,e)pyrene 二苯并(A,E)芘, Dibenzo(a,h)pyrene二苯并(A,H)芘; Dibenzo(a,i)pyrene二苯并(A,I)芘, Dibenzo(a,l)pyrene二苯并(A,L)芘, Benzo( e )pyrene二苯并(E)芘,1-Methylpyrene1-甲基芘Carcinogenic aryl amines 致癌性芳香胺(which are mentioned in the legal regulation for banned Azo colorants and which can be present respectively available also in free form; detectable at utilisation of the official legal test methods).pH value PH值Polyvinyl chloride (PVC) 聚氯乙烯Nonylphenol 壬基苯酚Octylphenol 辛基苯酚Nonylphenol-ethoxylate 壬基酚聚氧乙烯醚Lead – total content 含铅总量Tetrachlorophenols 四氯苯酚Trichlorophenols 三氯苯酚Tin organic compounds: 锡有机化合物TBT 三丁基锡(用于涂料和杀虫剂)TPhT 三苯基锡Tin organic compounds: 锡有机化合物MBT 2-硫醇基苯并噻唑(橡胶促进剂)DBT 二苯并噻吩(dibenzothiophene) DOT Chlorinated benzenes and toluenes 氯化苯和甲苯formamide 甲酰胺relevant at foamed materials (EVA), 乙烯醋酸乙烯酯共聚物 ethylene vinyl acetatefor instance in mats垫子, mattresses, seat contact surfaces 座椅接触面VOC (odour) 挥发性有机化合物(volatile organic compounds); at conspicuousnesses (screening)。
HSF-技术标准

物质:甲醛
对象禁止供货时期1 级各式使用于销售产品本体或其包装等用途,如扬声器、机架本体或栈板等木工产
品,或其他非木质材料制成的产品等。
立即禁止备注:栈板之甲醛限值不受本标准规范.
标准值(排放浓度):
胶合木质材料标准值(排放浓度):依木制品类型分二阶段管制排放浓度
说明:
第一阶段:Phase 1 (简称P1)
第二阶段:Phase 2 (简称P2)
HWPW-VC:Hardwood plywood—Veneer Core,单板芯之硬木胶合板
HWPW-CC:Hardwood plywood—Composite Core,复合芯之硬木胶合板
PB:ParticleBoard,粒片板
MDF:Medium density fiberboard,中密度纤维板
Thin MDF:最大厚度小于8mm 之MDF
(二) 其它非胶合木质材料排放浓度:
于12m3、1m3、或者0。
0225m3 之气密试验腔体中试验,于空气中的浓度在0。
1ppm (0。
124mg/m3)
以下(依照EN 717-1)
量测方法:
(一) 胶合木质材料:
(1)穿孔器测试法-EN 120 (PB,MDF, thin MDF)
(2)Chamber 测试法-ASTM E 1333-96(2002)
(3) 干燥器测试法-JIS A 1460
(二)其它非胶合木质材料:密闭腔体测试法-EN 717—1
甲醛︰不得检出。
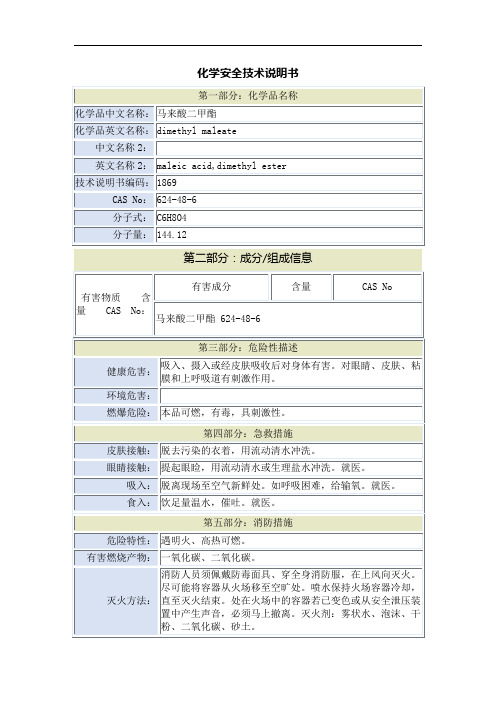
马来酸二甲酯安全技术说明书
操作注意事项:
密闭操作,注意通风。操作人员必须经过专门培训,严格遵守操作规程。建议操作人员佩戴自吸过滤式防毒面具(半面罩),戴化学安全防护眼镜,穿防毒物渗透工作服,戴橡胶耐油手套。远离火种、热源,工作场所严禁吸烟。使用防爆型的通风系统和设备。防止蒸气泄漏到工作场所空气中。避免与氧化剂、还原剂、酸类、碱类接触。搬运时要轻装轻卸,防止包装及容器损坏。配备相应品种和数量的消防器材及泄漏应急处理设备。倒空的容器可能残留有害物。
未制订标准
TLVWN:
未制订标准
工程控制:
密闭操作,注意通风。
呼吸系统防护:
空气中浓度超标时,必须佩戴自吸过滤式防毒面具(半面罩)。紧急事态抢救或撤离时,应该佩戴空气呼吸器。
眼睛防护:
戴化学安全防护眼镜。
身体防护:
穿防毒物渗透工作服。
手防护:
戴橡胶耐油手套。
其他防护:
工作现场严禁吸烟。工作完毕,淋浴更衣。特别注意眼和呼吸道的防护。
储存注意事项:
储存于阴凉、通风的库房。远离火种、热源。应与氧化剂、还原剂、酸类、碱类、食用化学品分开存放,切忌混储。配备相应品种和数量的消防器材。储区应备有泄漏应急处理设备和合适的收容材料。
第八部分:接触控制/个体防护
中国MAC(mg/m3):
未制定ቤተ መጻሕፍቲ ባይዱ准
前苏联MAC(mg/m3):
未制定标准
TLVTN:
第六部分:泄漏应急处理
应急处理:
迅速撤离泄漏污染区人员至安全区,并进行隔离,严格限制出入。切断火源。建议应急处理人员戴自给正压式呼吸器,穿防毒服。尽可能切断泄漏源。防止流入下水道、排洪沟等限制性空间。小量泄漏:用砂土、蛭石或其它惰性材料吸收。也可以用不燃性分散剂制成的乳液刷洗,洗液稀释后放入废水系统。大量泄漏:构筑围堤或挖坑收容。用泵转移至槽车或专用收集器内,回收或运至废物处理场所处置。
富马酸论文

前言前言富马酸二甲酯(Dimethyl Fumarat,简称DMF,化学名反丁烯二酸二甲酯),DMF 白色晶体,熔点为102~105,溶于乙醇/氯仿,微溶于水,常温下可以升华,对光和热稳定。
是新开发的一种优良的防腐保鲜剂,其效果优于苯甲酸、山梨酸、脱氢醋酸、丙酸盐等防腐剂。
因其具有低毒,高效,广泛抗菌特点,且对霉菌有特殊的抑制效果,可广泛用于食品,饲料,化妆品,水果,蔬菜及药物的防霉,防虫等,我国已批准作为食品防腐剂。
目前报道的能用于合成富马酸二甲酯的催化剂有很多种,传统的催化剂是浓H2SO4,但从富马酸二甲酯的生产成本、产品质量、催化剂的可再生重复使用以及环境保护这几个方面考虑,有必要寻求一种高效经济的催化剂,并将之用于目标产物的合成,要求这种催化剂能同时具有生产成本低、产品产率高、产品质量好、催化剂可再生重复使用、与产物易分离以及环境污染小等优点。
固体超强酸(Solid Superacid)是比酸度强于100%硫酸的酸。
固体超强酸的定义最初是由Gillespie给出的,即凡是固体酸强度大于100%硫酸的,都可称为固体超强酸。
由于固体表面性质的多样性、溶剂和指示剂在固体表面的强吸附作用等,使得用指示剂法测定的固体酸强度在应用中受到干扰。
同时,依靠肉眼观察颜色变化的判断方法也包含着某些不确定因素。
另一种常用的定义固体超强酸的方法,是以该固体为催化剂,在低于200℃的条件下,催化正丁烷异构化的活性来表示其酸强度。
这种表示方法具有很强的实用性而被采用。
最近,有人提出利用C-H和C-C键的键的碱性作为指示剂,固体超强酸使烷烃可逆质子化的能力表示其酸强度。
截止目前,已研制出许多种类、性能各异的固体超强酸。
大致可分为以下几类:(1)担载型,在高比表面固体表面担载液体超强酸SbF5等;(2)某些混合金属盐,如AlCl3,AlCl3-CuCl2,AlBr3,SbF3-FSO3H等;(3)硫酸根改性的金属氧化物,如SO42-/MxOy(M=Fe,Sn,Zr等);(4)复合金属化合物,如MO3/ZrO2,WO3/ZrO2等;(5)杂多酸,如H3PW12O40等;(6)氟化磺酸树脂,如H-Nafion等。
供应商DMF声明书(英文版-Sidney)

供应商DMF声明书SUPPLIER’S DMF DECLARATION丰叶电器制造厂(河源)有限公司:Fung Yip Electrical Manufacturing (He Y uan) Ltd. :作为贵公司的供应商,从本声明书签订之日起由我公司向贵公司供应的所有物料不含有富马酸二甲酯DMF,或者含有时在产品中的浓度不超过0.1mg/kg(0.1ppm),符合欧盟委员会《要求成员国保证含有生物杀虫剂富马酸二甲酯的产品不得投放市场和在市场上销售》,即2009/251/EC指令。
As one of your suppliers , this declaration verifies that all the materials supplied by our company fully do not contain the biocide dimethylfumarate,or the quantification limit of products should be 0.1mg DMF per kg or less from the dealaration authorization signature day on , our company ensures it fully complies with European Commission on the directive of《Requiring Member States to Ensure that Products Containing the Biocide Dimethylfumarate Are Not Placed or Made Available on the Market 》,viz 2009/251/EC DIRECTIVE .本保证书从签字盖章之日起生效。
The declaration becomes effective beginning on the day of signature stamp .供应商署名Supplier Signature。
富马酸FA_MSDS

富马酸含量分析毒性使用限量食品添加剂最大允许使用量最大允许残留量标准富马酸试剂级价格富马酸CAS号: 110-17-8英文名称: Fumaric acid英文同义词: FA;TMEDA;U-1149;fumaric;NSC-2752;FEMA2488;Fumarsure;boleticacid;FUMARSAEURE;Boleticacid中文名称: 富马酸中文同义词: 富马酸;紫堇酸;福馬酸;富馬酸;延胡索酸;別馬來酸;丁烯二酸;反丁烯二酸;反式紫堇酸;反丁烯二酸的CBNumber: CB5852804分子式: C4H4O4分子量: 116.07MOL File: 110-17-8.mol富马酸化学性质熔点: 298-300 °C (subl.)(lit.)密度: 1.62蒸气压: 1.7 mm Hg ( 165 °C)FEMA : 2488闪点: 230 °C水溶解性: 0.63 g/100 mL (25 ºC)Merck : 14,4287BRN : 605763稳定性: Stable at room temperature. Decomposes at around 230 C.Incompatible with strong oxidizing agents, bases, reducing agents.Combustible.CAS 数据110-17-8(CAS DataBase Reference)NIST化学物Fumaric acid(110-17-8)质信息:安全信息危险品标Xi志:危险类别36码:安全说明: 26危险品运输UN 9126编号:1WGKGermany :RTECS号: LS9625000毒害物质数110-17-8(Hazardous Substances Data)据:富马酸MSDS富马酸富马酸化学药品说明书复方草珊瑚含片—反丁烯二酸的测定—高效液相色谱法|药物分析方法信息富马酸性质、用途与生产工艺含量分析精确称取试样约1g,移入250ml锥形瓶中,加甲醇,50ml,在蒸汽浴上缓缓加热使试样溶解。
富马酸二甲酯介绍

富马酸二甲酯介绍富马酸二甲酯(Dimethylformamide,DMF)是一种有机化合物,化学式为C3H7NO,分子量为73.091 g/mol。
它是无色透明的液体,具有特殊的气味。
DMF在工业上广泛应用于溶剂、催化剂和反应中间体,同时也被用作纺织品、塑料、颜料等行业中的处理剂或添加剂。
下面将详细介绍富马酸二甲酯的性质、制备方法、应用以及安全注意事项。
1.物理性质:富马酸二甲酯是一种极性有机溶剂,其沸点为153-155°C,密度为0.944 g/cm³,闪点为57°C。
它与水混溶,可溶于大部分有机溶剂如乙醇、乙醚、苯等。
DMF在高温下具有可燃性,与空气形成易燃混合物。
此外,富马酸二甲酯还具有较高的介电常数和表面张力。
2.制备方法:(1)乙化胺法:将甲酰氨与乙烯胺在碱的催化下反应生成,然后再与甲醇反应制备DMF。
(2)二甲胺法:将甲醇与二甲胺在催化剂的作用下反应生成二甲甲酸酯,再与碳酸二乙酯反应后脱除乙酸生成DMF。
(3)甲酰氯法:将无水甲酸与氯化氢反应生成甲酰氯,再与甲胺发生酰胺化反应生成DMF。
3.应用:(1)富马酸二甲酯作为溶剂广泛应用于化学合成、有机合成和催化反应中,能与水、醇、酮、醚等多种溶剂混溶,具有良好的溶解性能。
(2)由于DMF极性较强,因此在染料、颜料、塑料等行业中作为溶剂使用时,能有效增加色料的分散性以及颜料和塑料的染色效果。
(3)由于富马酸二甲酯在很宽的温度范围内都有较好的稳定性,因此可用于高温工艺和高分子材料的制备。
(4)富马酸二甲酯还常被用于纺织行业中的维丝、涤纶纤维和氨纶纤维的溶解、纺丝和交联等工艺过程中,可以达到良好的效果。
4.安全注意事项:(1)DMF与空气中的氧气反应生成高温、易燃的混合物,因此在储存和使用时应避免与明火、静电火花等火源接触,并保持通风良好的工作环境。
(2)富马酸二甲酯经皮肤吸收性极强,对皮肤和黏膜有刺激作用,因此在使用时应注意佩戴防护手套、眼镜等器具,避免直接接触。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
________________________________________________________________ _____________ ___________________ ___________________HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use TECFIDERA safely and effectively. See full prescribing information for TECFIDERA.TECFIDERA® (dimethyl fumarate) delayed-release capsules, for oral use Initial U.S. Approval: 2013RECENT MAJOR CHANGESDosage and Administration, Dosing Information (2.1) 12/2014 Contraindications (4) 12/2014 Warnings and Precautions, Anaphylaxis and Angioedema (5.1) 12/2014 Warnings and Precautions, PML (5.2) 12/2014 Warnings and Precautions, Lymphopenia (5.3) 12/2014INDICATIONS AND USAGETECFIDERA is indicated for the treatment of patients with relapsing forms of multiple sclerosis (1)DOSAGE AND ADMINISTRATION• Starting dose: 120 mg twice a day, orally, for 7 days (2.1)• Maintenance dose after 7 days: 240 mg twice a day, orally (2.1)• Swallow TECFIDERA capsules whole and intact. Do not crush, chew, or sprinkle capsule contents on food (2.1)• Take TECFIDERA with or without food (2.1)DOSAGE FORMS AND STRENGTHSDelayed-release capsules: 120 mg and 240 mg (3)Tecfidera NDA 204063/S-003, S-008, S-010FDA Approved Labeling Text 12/3/14CONTRAINDICATIONSKnown hypersensitivity to dimethyl fumarate or any of the excipients of TECFIDERA. (4)WARNINGS AND PRECAUTIONS _______________ • Anaphylaxis and angioedema: Discontinue and do not restart TECFIDERA if these occur. (5.1)• Progressive multifocal leukoencephalopathy (PML): Withhold TECFIDERA at the first sign or symptom suggestive of PML. (5.2)• Lymphopenia: Obtain a CBC including lymphocyte count before initiating TECFIDERA, after 6 months, and every 6 to 12 months thereafter.Consider interruption of TECFIDERA if lymphocyte counts <0.5 x 109/L persist for more than six months. (5.3)ADVERSE REACTIONSMost common adverse reac tions (incidence ≥10% and ≥2% placebo) were flushing, abdominal pain, diarrhea, and nausea. (6.1)To report SUSPECTED ADVERSE REACTIONS, contact Biogen Idec at 1-800-456-2255 or FDA at 1-800-FDA-1088 or /medwatch.USE IN SPECIFIC POPULATIONSPregnancy: based on animal data, may cause fetal harm. (8.1)See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labelingRevised: 12/2014FULL PRESCRIBING INFORMATION: CONTENTS*1 INDICATIONS AND USAGE2 DOSAGE AND ADMINISTRATION2.1. Dosing Information2.2 Blood Test Prior to Initiation of Therapy3 DOSAGE FORMS AND STRENGTHS4 CONTRAINDICATIONS5 WARNINGS AND PRECAUTIONS5.1 Anaphylaxis and Angioedema5.2 Progressive Multifocal Leukoencephalopathy5.3 Lymphopenia5.4 Flushing6 ADVERSE REACTIONS6.1 Clinical Trials Experience8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy8.3 Nursing Mothers8.4 Pediatric Use8.5 Geriatric Use11 DESCRIPTION12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.2 Pharmacodynamics12.3 Pharmacokinetics13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility13.2 Animal Toxicology and/or Pharmacology14 CLINICAL STUDIES16 HOW SUPPLIED/STORAGE AND HANDLING17 PATIENT COUNSELING INFORMATION*Sections or subsections omitted from the full prescribing information are not listed.FULL PRESCRIBING INFORMATION1 INDICATIONS AND USAGETECFIDERA is indicated for the treatment of patients with relapsing forms of multiple sclerosis.2 DOSAGE AND ADMINISTRATION2.1 Dosing InformationThe starting dose for TECFIDERA is 120 mg twice a day orally. After 7 days, the dose should be increased to the maintenance dose of 240 mg twice a day orally. TECFIDERA should be swallowed whole and intact. TECFIDERA should not be crushed or chewed and the capsule contents should not be sprinkled on food. TECFIDERA can be taken with or without food. Temporary dose reductions to 120 mg twice a day may be considered for individuals who do not tolerate the maintenance dose. Within 4 weeks, the recommended dose of 240 mg twice a day should be resumed. Discontinuation of TECFIDERA should be considered for patients unable to tolerate return to the maintenance dose. The incidence of flushing may be reduced by administration of TECFIDERA with food. Alternatively, administration of non-enteric coated aspirin (up to a dose of 325 mg) 30 minutes prior to TECFIDERA dosing may reduce the incidence or severity of flushing [see Clinical Pharmacology (12.3)].2.2 Blood Test Prior to Initiation of TherapyObtain a complete blood cell count (CBC) including lymphocyte count before initiation of therapy [see Warnings and Precautions (5.3)].3 DOSAGE FORMS AND STRENGTHSTECFIDERA is available as hard gelatin delayed-release capsules containing 120 mg or 240 mg of dimethyl fum arate. The 120 mg capsules have a green cap and white body, printed with “BG 12 120 mg” in black ink on the body. The 240 mg capsules have a green cap and a green body, printed with “BG-12 240 mg” in black ink on the body.4 CONTRAINDICATIONSTECFIDERA is contraindicated in patients with known hypersensitivity to dimethyl fumarate or to any of the excipients of TECFIDERA. Reactions have included anaphylaxis and angioedema [see Warnings and Precautions (5.1)].5 WARNINGS AND PRECAUTIONS5.1 Anaphylaxis and AngioedemaTECFIDERA can cause anaphylaxis and angioedema after the first dose or at any time during treatment. Signs and symptoms have included difficulty breathing, urticaria, and swelling of the throat and tongue. Patients should be instructed to discontinue TECFIDERA and seek immediate medical care should they experience signs and symptoms of anaphylaxis or angioedema.5.2 Progressive Multifocal LeukoencephalopathyA fatal case of progressive multifocal leukoencephalopathy (PML) occurred in a patient with MS who received TECFIDERA for 4 years while enrolled in a clinical trial. PML is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically only occurs in patients who are immunocompromised, and that usually leads to death or severe disability. During the clinical trial, the patient experienced prolonged lymphopenia (lymphocyte counts predominantly <0.5x109/L for 3.5 years) while taking TECFIDERA [see Warnings and Precautions (5.3)]. The role of lymphopenia in this case is unknown. The patient had no other identified systemic medical conditions resulting in compromised immune system function and had not previously been treated with natalizumab, which has a known association with PML. The patient was also not taking any immunosuppressive or immunomodulatory medications concomitantly.At the first sign or symptom suggestive of PML, withhold TECFIDERA and perform an appropriate diagnostic evaluation. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.5.3 LymphopeniaTECFIDERA may decrease lymphocyte counts. In the MS placebo controlled trials, mean lymphocyte counts decreased by approximately 30% during the first year of treatment with TECFIDERA and then remained stable. Four weeks after stopping TECFIDERA, mean lymphocyte counts increased but did not return to baseline. Six percent (6%) of TECFIDERA patients and <1% of placebo patients experienced lymphocyte counts <0.5x109/L (lower limit of normal 0.91x109/L). The incidence of infections (60% vs 58%) and serious infections (2% vs 2%) was similar in patients treated with TECFIDERA or placebo, respectively. There was no increased incidence of serious infections observed in patients with lymphocyte counts<0.8x109/L or 0.5x109/L in controlled trials, although one patient in an extension study developed PML in the setting of prolonged lymphopenia (lymphocyte counts predominantly<0.5x109/L for 3.5 years) [see Warnings and Precautions (5.2)]. In controlled and uncontrolled clinical trials, 2% of patients experienced lymphocyte counts <0.5 x 109/L for at least six months. In these patients, the majority of lymphocyte counts remained <0.5x109/L with continued therapy. TECFIDERA has not been studied in patients with pre-existing low lymphocyte counts. Before initiating treatment with TECFIDERA, a CBC including lymphocyte count should be obtained. A CBC including lymphocyte count should also be obtained after 6 months of treatment, every 6 to 12 months thereafter, and as clinically indicated. Consider interruption ofTECFIDERA in patients with lymphocyte counts <0.5 x 109/L persisting for more than six months. Given the potential for delay in lymphocyte recovery after discontinuation of TECFIDERA, consider following lymphocyte counts until lymphopenia is resolved. Withholding treatment should be considered in patients with serious infections until the infection(s) is resolved. Decisions about whether or not to restart TECFIDERA should be individualized based on clinical circumstances.5.4 FlushingTECFIDERA may cause flushing (e.g., warmth, redness, itching, and/or burning sensation). In clinical trials, 40% of TECFIDERA treated patients experienced flushing. Flushing symptoms generally began soon after initiating TECFIDERA and usually improved or resolved over time. In the majority of patients who experienced flushing, it was mild or moderate in severity. Three percent (3%) of patients discontinued TECFIDERA for flushing and <1% had serious flushing symptoms that were not life-threatening but led to hospitalization. Administration of TECFIDERA with food may reduce the incidence of flushing. Alternatively, administration of non-enteric coated aspirin (up to a dose of 325 mg) 30 minutes prior to TECFIDERA dosing may reduce the incidence or severity of flushing [see Dosing and Administration (2.1) and Clinical Pharmacology (12.3)].6 ADVERSE REACTIONSThe following important adverse reactions are described elsewhere in labeling: • Anaphylaxis and Angioedema [see Warnings and Precautions (5.1)].• Progressive multifocal leukoencephalopathy [see Warnings and Precautions (5.2)] .• Lymphopenia [see Warnings and Precautions (5.3)].• Flushing [see Warnings and Precautions (5.4)].6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.The most common adverse reactions (incidence ≥10% and ≥2% more than placebo) for TECFIDERA were flushing, abdominal pain, diarrhea, and nausea.Adverse Reactions in Placebo-Controlled TrialsIn the two well-controlled studies demonstrating effectiveness, 1529 patients received TECFIDERA with an overall exposure of 2244 person-years [see Clinical Studies (14)].The adverse reactions presented in the table below are based on safety information from 769 patients treated with TECFIDERA 240 mg twice a day and 771 placebo-treated patients.Table 1: Adverse Reactions in Study 1 and 2 reported for TECFIDERA 240 mg BID at ≥ 2% higher incidence than placeboTECFIDERA PlaceboN=769 N=771% %GastrointestinalTECFIDERA caused GI events (e.g., nausea, vomiting, diarrhea, abdominal pain, and dyspepsia). The incidence of GI events was higher early in the course of treatment (primarily in month 1) and usually decreased over time in patients treated with TECFIDERA compared with placebo. Four percent (4%) of patients treated with TECFIDERA and less than 1% of placebo patients discontinued due to gastrointestinal events. The incidence of serious GI events was 1% in patients treated with TECFIDERA.Hepatic TransaminasesAn increased incidence of elevations of hepatic transaminases in patients treated with TECFIDERA was seen primarily during the first six months of treatment, and most patients with elevations had levels < 3 times the upper limit of normal (ULN). Elevations of alanine aminotransferase and aspartate aminotransferase to ≥ 3 times the ULN occurred in a small number of patients treated with both TECFIDERA and placebo and were balanced betweengro ups. There were no elevations in transaminases ≥ 3 times the ULN with concomitant elevations in total bilirubin > 2 times the ULN. Discontinuations due to elevated hepatic transaminases were < 1% and were similar in patients treated with TECFIDERA or placebo. EosinophiliaA transient increase in mean eosinophil counts was seen during the first 2 months of therapy. Adverse Reactions in Placebo-Controlled and Uncontrolled StudiesIn placebo-controlled and uncontrolled clinical studies, a total of 2513 patients have received TECFIDERA and been followed for periods up to 4 years with an overall exposure of 4603 person-years. Approximately 1162 patients have received more than 2 years of treatment with TECFIDERA. The adverse reaction profile of TECFIDERA in the uncontrolled clinical studies was consistent with the experience in the placebo-controlled clinical trials.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category CThere are no adequate and well-controlled studies in pregnant women. In animals, adverse effects on offspring survival, growth, sexual maturation, and neurobehavioral function were observed when dimethyl fumarate (DMF) was administered during pregnancy and lactation at clinically relevant doses. TECFIDERA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.In rats administered DMF orally (25, 100, 250 mg/kg/day) throughout organogenesis, embryofetal toxicity (reduced fetal body weight and delayed ossification) were observed at the highest dose tested. This dose also produced evidence of maternal toxicity (reduced body weight). Plasma exposure (AUC) for monomethyl fumarate (MMF), the major circulating metabolite, at the no-effect dose is approximately three times that in humans at the recommended human dose (RHD) of 480 mg/day. In rabbits administered DMF orally (25, 75, and 150mg/kg/day) throughout organogenesis, embryolethality and decreased maternal body weight were observed at the highest dose tested. The plasma AUC for MMF at the no-effect dose is approximately 5 times that in humans at the RHD.Oral administration of DMF (25, 100, and 250 mg/kg/day) to rats throughout organogenesis and lactation resulted in increased lethality, persistent reductions in body weight, delayed sexual maturation (male and female pups), and reduced testicular weight at the highest dose tested. Neurobehavioral impairment was observed at all doses. A no-effect dose for developmental toxicity was not identified. The lowest dose tested was associated with plasma AUC for MMF lower than that in humans at the RHD.Pregnancy RegistryThere is a pregnancy registry that monitors pregnancy outcomes in women exposed to TECFIDERA during pregnancy. Encourage patients to enroll by calling 1-866-810-1462 or visiting .8.3 Nursing MothersIt is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when TECFIDERA is administered to a nursing woman.8.4 Pediatric UseSafety and effectiveness in pediatric patients have not been established.8.5 Geriatric UseClinical studies of TECFIDERA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.11 DESCRIPTION TECFIDERA contains dimethyl fumarate which is also known by its chemical name, dimethyl(E) butenedioate, (C 6H 8O 4). It has the following structure: HHOOODimethyl fumarate is a white to off-white powder that is highly soluble in water with a molecular mass of 144.13. TECFIDERA is provided as hard gelatin delayed-release capsules for oral administration, containing 120 mg or 240 mg of dimethyl fumarate consisting of the following inactive ingredients: microcrystalline cellulose, silicified microcrystalline cellulose, croscarmellose sodium, talc, silica colloidal silicon dioxide, magnesium stearate, triethyl citrate, methacrylic acid copolymer -Type A, methacrylic acid copolymer dispersion, simethicone (30% emulsion),sodium lauryl sulphate, and polysorbate 80. The capsule shell, printed with black ink, contains the following inactive ingredients: gelatin, titanium dioxide, FD&C blue 1; brilliant blue FCF, yellow iron oxide and black iron oxide. 12 CLINICAL PHARMACOLOGY 12.1 Mechanism of Action The mechanism by which dimethyl fumarate (DMF) exerts its therapeutic effect in multiple sclerosis is unknown. DMF and the metabolite, monomethyl fumarate (MMF), have been shown to activate the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway in vitro and in vivo in animals and humans. The Nrf2 pathway is involved in the cellular response to oxidative stress. MMF has been identified as a nicotinic acid receptor agonist in vitro . 12.2 Pharmacodynamics Potential to prolong the QT interval In a placebo controlled thorough QT study performed in healthy subjects, there was no evidence that dimethyl fumarate caused QT interval prolongation of clinical significance (i.e., the upper bound of the 90% confidence interval for the largest placebo-adjusted, baseline-corrected QTc was below 10 ms).12.3 PharmacokineticsAfter oral administration of TECFIDERA, dimethyl fumarate undergoes rapid presystemic hydrolysis by esterases and is converted to its active metabolite, monomethyl fumarate (MMF). Dimethyl fumarate is not quantifiable in plasma following oral administration of TECFIDERA. Therefore all pharmacokinetic analyses related to TECFIDERA were performed with plasma MMF concentrations. Pharmacokinetic data were obtained in subjects with multiple sclerosis and healthy volunteers.AbsorptionThe median T max of MMF is 2-2.5 hours. The peak plasma concentration (C max) and overall exposure (AUC) increased approximately dose proportionally in the dose range studied (120 mg to 360 mg). Following administration of TECFIDERA 240 mg twice a day with food, the mean C max of MMF was 1.87 mg/L and AUC was 8.21 mg.hr/L in MS patients.A high-fat, high-calorie meal did not affect the AUC of MMF but decreased its Cmax by 40%. The T max was delayed from 2.0 hours to 5.5 hours. In this study, the incidence of flushing was reduced by approximately 25% in the fed state.DistributionThe apparent volume of distribution of MMF varies between 53 and 73 L in healthy subjects. Human plasma protein binding of MMF is 27-45% and independent of concentration. MetabolismIn humans, dimethyl fumarate is extensively metabolized by esterases, which are ubiquitous in the gastrointestinal tract, blood, and tissues, before it reaches the systemic circulation. Further metabolism of MMF occurs through the tricarboxylic acid (TCA) cycle, with no involvement of the cytochrome P450 (CYP) system. MMF, fumaric and citric acid, and glucose are the major metabolites in plasma.EliminationExhalation of CO2 is the primary route of elimination, accounting for approximately 60% of the TECFIDERA dose. Renal and fecal elimination are minor routes of elimination, accounting for 16% and 1% of the dose respectively. Trace amounts of unchanged MMF were present in urine. The terminal half-life of MMF is approximately 1 hour and no circulating MMF is present at 24 hours in the majority of individuals. Accumulation of MMF does not occur with multiple doses of TECFIDERA.Specific PopulationsBody weight, gender, and age do not require dosage adjustment.No studies have been conducted in subjects with hepatic or renal impairment. However, neither condition would be expected to affect exposure to MMF and therefore no dosage adjustment is necessary.Drug Interaction StudiesNo potential drug interactions with dimethyl fumarate or MMF were identified in in vitro CYP inhibition and induction studies, or in P-glycoprotein studies. Single doses of interferon beta-1a or glatiramer acetate did not alter the pharmacokinetics of MMF. Aspirin, when administered approximately 30 minutes before TECFIDERA, did not alter the pharmacokinetics of MMF.13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility CarcinogenesisCarcinogenicity studies of dimethyl fumarate (DMF) were conducted in mice and rats. In mice, oral administration of DMF (25, 75, 200, and 400 mg/kg/day) for up to two years resulted in an increase in nonglandular stomach (forestomach) and kidney tumors: squamous cell carcinomas and papillomas of the forestomach in males and females at 200 and 400 mg/kg/day; leiomyosarcomas of the forestomach at 400 mg/kg/day in males and females; renal tubular adenomas and carcinomas at 200 and 400 mg/kg/day in males; and renal tubule adenomas at 400 mg/kg/day in females. Plasma MMF exposure (AUC) at the highest dose not associated with tumors in mice (75 mg/kg/day) was similar to that in humans at the recommended human dose (RHD) of 480 mg/day.In rats, oral administration of DMF (25, 50, 100, and 150 mg/kg/day) for up to two years resulted in increases in squamous cell carcinomas and papillomas of the forestomach at all doses tested in males and females, and in testicular interstitial (Leydig) cell adenomas at 100 and 150mg/kg/day. Plasma MMF AUC at the lowest dose tested was lower than that in humans at the RHD.MutagenesisDimethyl fumarate (DMF) and monomethyl fumarate (MMF) were not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. DMF and MMF were clastogenic in the in vitro chromosomal aberration assay in human peripheral blood lymphocytes in the absence of metabolic activation. DMF was not clastogenic in the in vivo micronucleus assay in the rat. Impairment of FertilityIn male rats, oral administration of DMF (75, 250, and 375 mg/kg/day) prior to and throughout the mating period had no effect on fertility; however, increases in non-motile sperm were observed at the mid and high doses. The no-effect dose for adverse effects on sperm is similar to the recommended human dose (RHD) of 480 mg/day on a body surface area (mg/m2) basis.In female rats, oral administration of DMF (20, 100, and 250 mg/kg/day) prior to and during mating and continuing to gestation day 7 caused disruption of the estrous cycle and increases in embryolethality at the highest dose tested. The highest dose not associated with adverse effects (100 mg/kg/day) is twice the RHD on a mg/m2 basis.Testicular toxicity (germinal epithelial degeneration, atrophy, hypospermia, and/or hyperplasia) was observed at clinically relevant doses in mice, rats, and dogs in subchronic and chronic oral toxicity studies of DMF, and in a chronic oral toxicity study evaluating a combination of four fumaric acid esters (including DMF) in rats.13.2 Animal Toxicology and/or PharmacologyKidney toxicity was observed after repeated oral administration of dimethyl fumarate (DMF) in mice, rats, dogs, and monkeys. Renal tubule epithelia regeneration, suggestive of tubule epithelial injury, was observed in all species. Renal tubular hyperplasia was observed in rats with dosing for up to two years. Cortical atrophy and interstitial fibrosis were observed in dogs and monkeys at doses above 5 mg/kg/day. In monkeys, the highest dose tested (75 mg/kg/day) was associated with single cell necrosis and multifocal and diffuse interstitial fibrosis, indicating irreversible loss of renal tissue and function. In dogs and monkeys, the 5 mg/kg/day dose was associated with plasma MMF exposures less than or similar to that in humans at the recommended human dose (RHD).A dose-related increase in incidence and severity of retinal degeneration was observed in mice following oral administration of DMF for up to two years at doses above 75 mg/kg/day, a dose associated with plasma MMF exposure (AUC) similar to that in humans at the RHD.14 CLINICAL STUDIESThe efficacy and safety of TECFIDERA were demonstrated in two studies (Studies 1 and 2) that evaluated TECFIDERA taken either twice or three times a day in patients with relapsing-remitting multiple sclerosis (RRMS). The starting dose for TECFIDERA was 120 mg twice or three times a day for the first 7 days, followed by an increase to 240 mg twice or three times a day. Both studies included patients who had experienced at least 1 relapse over the year preceding the trial or had a brain Magnetic Resonance Imaging (MRI) scan demonstrating at least one gadolinium-enhancing (Gd+) lesion within 6 weeks of randomization. The Expanded Disability Status Scale (EDSS) was also assessed and patients could have scores ranging from 0 to 5. Neurological evaluations were performed at baseline, every 3 months, and at the time of suspected relapse. MRI evaluations were performed at baseline, month 6, and year 1 and 2 in a subset of patients (44% in Study 1 and 48% in Study 2).Study 1: Placebo-Controlled Trial in RRMSStudy 1 was a 2-year randomized, double-blind, placebo-controlled study in 1234 patients with RRMS. The primary endpoint was the proportion of patients relapsed at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of new T1 hypointense lesions, number of Gd+ lesions, annualized relapse rate (ARR), and time to confirmed disability progression. Confirmed disability progression was defined as at least a 1 point increase from baseline EDSS (1.5 point increase for patients with baseline EDSS of 0) sustained for 12 weeks.Patients were randomized to receive TECFIDERA 240 mg twice a day (n=410), TECFIDERA 240 mg three times a day (n=416), or placebo (n=408) for up to 2 years. The median age was 39 years, median time since diagnosis was 4 years, and median EDSS score at baseline was 2. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 69% for patients assigned to TECFIDERA 240 mg twice a day, 69% for patients assigned to TECFIDERA 240 mg three times a day and 65% for patients assigned to placebo groups.TECFIDERA had a statistically significant effect on all of the endpoints described above and the 240 mg three times daily dose showed no additional benefit over the TECFIDERA 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 2 and Figure 1.Table 2: Clinical and MRI Results of Study 1Relative risk reduction 49%Figure 1: Time to 12-Week Confirmed Progression of Disability (Study 1)Study 2: Placebo-Controlled Trial in RRMSStudy 2 was a 2-year multicenter, randomized, double-blind, placebo-controlled study that also included an open-label comparator arm in patients with RRMS. The primary endpoint was the annualized relapse rate at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of T1 hypointense lesions, number of Gd+ lesions, proportion of patients relapsed, and time to confirmed disability progression as defined in Study 1.Patients were randomized to receive TECFIDERA 240 mg twice a day (n=359), TECFIDERA 240 mg three times a day (n=345), an open-label comparator (n=350), or placebo (n=363) for up to 2 years. The median age was 37 years, median time since diagnosis was 3 years, and median EDSS score at baseline was 2.5. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 70% for patients assigned to TECFIDERA 240 mg twice a day, 72% for patients assigned to TECFIDERA 240 mg three times a day and 64% for patients assigned to placebo groups.。