计算材料学答案
材料科学基础习题与参考答案(doc14页)完美版

材料科学基础习题与参考答案(doc14页)完美版第⼀章材料的结构⼀、解释以下基本概念空间点阵、晶格、晶胞、配位数、致密度、共价键、离⼦键、⾦属键、组元、合⾦、相、固溶体、中间相、间隙固溶体、置换固溶体、固溶强化、第⼆相强化。
⼆、填空题1、材料的键合⽅式有四类,分别是(),(),(),()。
2、⾦属原⼦的特点是最外层电⼦数(),且与原⼦核引⼒(),因此这些电⼦极容易脱离原⼦核的束缚⽽变成()。
3、我们把原⼦在物质内部呈()排列的固体物质称为晶体,晶体物质具有以下三个特点,分别是(),(),()。
4、三种常见的⾦属晶格分别为(),()和()。
5、体⼼⽴⽅晶格中,晶胞原⼦数为(),原⼦半径与晶格常数的关系为(),配位数是(),致密度是(),密排晶向为(),密排晶⾯为(),晶胞中⼋⾯体间隙个数为(),四⾯体间隙个数为(),具有体⼼⽴⽅晶格的常见⾦属有()。
6、⾯⼼⽴⽅晶格中,晶胞原⼦数为(),原⼦半径与晶格常数的关系为(),配位数是(),致密度是(),密排晶向为(),密排晶⾯为(),晶胞中⼋⾯体间隙个数为(),四⾯体间隙个数为(),具有⾯⼼⽴⽅晶格的常见⾦属有()。
7、密排六⽅晶格中,晶胞原⼦数为(),原⼦半径与晶格常数的关系为(),配位数是(),致密度是(),密排晶向为(),密排晶⾯为(),具有密排六⽅晶格的常见⾦属有()。
8、合⾦的相结构分为两⼤类,分别是()和()。
9、固溶体按照溶质原⼦在晶格中所占的位置分为()和(),按照固溶度分为()和(),按照溶质原⼦与溶剂原⼦相对分布分为()和()。
10、影响固溶体结构形式和溶解度的因素主要有()、()、()、()。
11、⾦属化合物(中间相)分为以下四类,分别是(),(),(),()。
12、⾦属化合物(中间相)的性能特点是:熔点()、硬度()、脆性(),因此在合⾦中不作为()相,⽽是少量存在起到第⼆相()作⽤。
13、CuZn、Cu5Zn8、Cu3Sn的电⼦浓度分别为(),(),()。
材料科学基础习题参考答案.docx

材料科学基础习题参考答案 第一章材料结构的基本知识8.计算下列晶体的离于键与共价键的相对比例。
(1) NaF (2) CaO (3) ZnS解:(1)查表得:X Na =0.93,X F =3.98--(0.93-3.98)2根据鲍林公式可得NaF 中离子键比例为:[1-e 4 ]x 100% = 90.2%共价键比例为:1-90.2%=9.8%--(1.00-3.44 )2(2) 同理,CaO 中离子键比例为:[1-e 4 ]x 100% = 77.4%共价键比例为:1-77.4%=22.6%(3) ZnS 中离子键比例为:Z“S 中离子键含量=[1 -£-1/4'2-58-165)2]x 100% = 19.44% 共价键比例为:1-19.44%=80.56%10说明结构转变的热力学条件与动力学条件的意义.说明稳态结构与亚稳态结构之间的关 系。
答:结构转变的热力学条件决定转变是否可行,是结构转变的推动力,是转变的必要条件; 动力学条件决定转变速度的大小,反映转变过程中阻力的大小。
稳态结构与亚稳态结构之间的关系:两种状态都是物质存在的状态,材料得到的结构是 稳态或亚稳态,取决于转变过程的推动力和阻力(即热力学条件和动力学条件),阻力小时得 到稳态结构,阻力很大时则得到亚稳态结构。
稳态结构能量最低,热力学上最稳定;亚稳态 结构能量高,热力学上不稳定,但向稳定结构转变速度慢,能保持相对稳定甚至长期存在。
但在一定条件下,亚稳态结构向稳态结构转变。
1.第二章九材料中的騒須勾)与[2廊1)与[112], (110)与[111], (132)与[123], (322)与[236]指数。
题: 系的 (21 在立方晶系的一个晶胞虫画出(111丄和丄112、日面.才晶系的画出同M1)、■'朋两晶面交钱亠 1]晶向。
112) d2. 有一正交点阵的a=b, c=a/2o 某晶面在三个晶轴上的截距分别为6个、2个和4个原子 间距,求该晶面的密勒指数。
计算材料学之蒙特卡洛方法论述

计算材料学之蒙特卡洛方法一、计算材料学要紧内容计算材料学涉及材料的各个方面,如不同层次的结构、各种性能等等,因此,有专门多相应的计算方法。
在进行材料计算时,首先要依照所要计算的对象、条件、要求等因素选择适当的方法。
要想做好选择,必须了解材料计算方法的分类。
目前,要紧有两种分类方法:一是按理论模型和方法分类,二是按材料计算的特征空间尺寸(Characteristic space scale)分类。
材料的性能在专门大程度上取决于材料的微结构,材料的用途不同,决定其性能的微结构尺度会有专门大的差不。
例如,对结构材料来讲,阻碍其力学性能的结构尺度在微米以上,而关于电、光、磁等功能材料来讲可能要小到纳米,甚至是电子结构。
因此,计算材料学的研究对象的特征空间尺度从埃到米。
时刻是计算材料学的另一个重要的参量。
关于不同的研究对象或计算方法,材料计算的时刻尺度可从10-15秒(如分子动力学方法等)到年(如关于腐蚀、罐变、疲劳等的模拟)。
关于具有不同特征空间、时刻尺度的研究对象,均有相应的材料计算方法。
目前常用的计算方法包括第一原理从头计算法,分子动力学方法,蒙特卡洛方法,有限元分析等。
下面要紧介绍蒙特卡罗方法:蒙特卡罗方法:一、方法的简介蒙特•卡罗方法(Monte Carlo method),也称统计模拟方法,是二十世纪四十年代中期由于科学技术的进展和电子计算机的发明,而被提出的一种以概率统计理论为指导的一类特不重要的数值计算方法。
是指使用随机数(或更常见的伪随机数)来解决专门多计算问题的方法。
与它对应的是确定性算法这种方法作为一种独立的方法被提出来,并首先在核武器的试验与研制中得到了应用。
蒙特卡罗方法是一种计算方法,但与一般数值计算方法有专门大区不。
它是以概率统计理论为基础的一种方法。
由于蒙特卡罗方法能够比较逼真地描述事物的特点及物理实验过程,解决一些数值方法难以解决的问题,因而该方法的应用领域日趋广泛。
蒙特•卡罗方法在金融工程学,宏观经济学,计算物理学(如粒子输运计算、量子热力学计算、空气动力学计算)等领域应用广泛。
计算材料学总复习A

电子在一个等效的中心势场中的运动,等效势场包括原子核对该电子作用势
和其他电子对该电子的平均作用势。
24、原子中单电子波函数 φi(qi)称为自旋轨道。描述原子中单电子空间运动状态
的波函数ϕ i
(rri
)
称为原子轨道。
25、将分子轨道表示成原子轨道线性组合的方法,称为原子轨道线性组合—分子
轨道法(LCAO-MO 方法)。
∆Z ∆E
其中 ∆Z 表示能量在 E ~ (E + ∆E)范围内的电子能态数目。 32、费米能级又称为费米能,是遵从泡利不相容原理的电子体系的化学势。从费
米狄拉克分布函数上看,费米能级在数值上等于电子占居几率为 1/2 的量子 态的能量。在绝对零度(0 K )时,电子均按泡利不相容原理填充于能量低于费 米能的状态中,即在费米能级以下的状态全部是满的,而费米能级以上的状
并利用 Hartree-Fock 近似将多电子问题变为单电子问题,用 Hartree-Fock
自洽场方法或密度泛函理论求解单电子问题。建立在 Hohenberg-Kohn 定理
之上密度泛函理论是目前求解单电子问题的最精确的理论。
23、对多电子原子的计算主要采用单电子近似,即把每个电子的运动近似成单个
B 正则系综
C 等温等压系综
D 等焓等压系综
2、在热力学统计物理中该系综是一个粒子数为 N,体积为 V,温度为 T 和总动
量为守恒量的系综,在这个系综中系统的粒子数(N)、体积(V)和温度(T)都保
持不变,并且总动量为零,因此为称( B )系综。(√)
A 微正则系综
B 正则系综
C 等温等压系综
D 等焓等压系综
态全部是空的。
33、固体能带计算就是求解对固体系统做了三个近似后得到的单电子方程,对这
计算材料学答案

计算材料学1.计算材料学在不同层次使用的计算方法2.材料科学与工程的四要素包括材料固有性质、结构与组分、使用性能、合成与加工。
3.根据晶体中原子间的结合力类型划分有哪几种晶体类型,其特点是什么?类型作用力键合特点离子键静电库仑引力最强无方向性、配位数高、熔点高、强度高、低膨胀系数、塑性差、导电性差、溶态导电共价键轨道相互作用强有方向性、配位数低、熔点高、强度高、低膨胀系数、塑性差、溶态不导电金属键原子实与自由电子之间的景点相互作用(库仑力)较强无方向性,配位数高,塑性好,导电性好,导热性好,结构密堆分子键瞬时偶极矩较弱无方向性,配位数高,结构密堆,熔点高,绝缘氢键氢原子核与极性分子间的库仑力弱无方向性,无饱和性当样品从绝对零度开始加热时,只有能量位于费米能附近kBT 范围内的轨道电子才被热激发,每个被激发电子所获得的能量量级正好为 kBT , 所以被激发电子的比例约为 T/TF ,设 N 为电子总数,则被激发的电子数约为 NT/TF ,在被激发的 NT/TF 个电子中,每个都具有能级为 kBT 的热能,所以总的电子热能的量级为,于是电子热容为,正比于温度 T ,与实验结果一致。
室温下, TF 约为 5×104 K ,Cel 比经典值 (3/2)NkB 约小两个量级。
5. 自由电子轮的两个假设(1) 假设自由电子在金属晶体中的恒定势场下运动(2) 单电子近似 6. 根据,T 的实验数据作图求出电子气的比热容常数()Tk T T NU FB ~7.自由电子近似与近自由电子近似的主要区别自由电子近似(遵循波尔兹曼统计规律):假设电子在金属晶体的恒定场中运动,在此模型下建立薛定谔方程;单电子近似,即自由电子理论把电子的运动状态从一个多体问题转化为单体问题处理,处理过程中考虑电子费米对称性。
近自由电子近似(遵循费米-狄拉克统计):除了自由电子近似的两个假设外,还大胆的做出如下近似(1)绝热近似:原子核(或离子实)的质量>>电子质量,离子运动速度慢,假设离子固定在瞬时位置上,多体问题化成多电子问题(2)自洽场(H-F)方法:多电子问题单电子问题,每个电子是在固定的离子势场及其它电子的平均场中运动(3)认为所有离子势场和其他电子的平均场是周期性势场。
(完整版)材料科学基础经典习题及答案(20210128050106)

第一章1. 作图表示立方晶体的123,012,421晶面及102,211,346晶向。
2. 在六方晶体中,绘出以下常见晶向0001,2110,1010,1120,1210等。
3. 写出立方晶体中晶面族{100},{110},{111},{112}等所包括的等价晶面。
4. 镁的原子堆积密度和所有hep金属一样,为0.74。
试求镁单位晶3胞的体积。
已知Mg的密度m g 1.74Mg/m,相对原子质量为24.31,原子半径r=0.161 nm。
5. 当CN=6寸Na离子半径为0.097nm,试问:1)当CN=40寸,其半径为多少?2)当CN=8时,其半径为多少?6. 试问:在铜(fcc,a=0.361 nm)的<100>方向及铁(bcc,a=0.286nm)的<100>方向,原子的线密度为多少?7. 镍为面心立方结构,其原子半径为r Ni°・1246nm。
试确定在镍的(100),(110)及(111)平面上1 mm2中各有多少个原子。
8. 石英Si°2的密度为2.65 Mg/m。
试问:1) 1 m3中有多少个硅原子(与氧原子)?2)当硅与氧的半径分别为0.038nm与0.114nm时,其堆积密度为多少(假设原子是球形的)?109. 在800C时10个原子中有一个原子具有足够能量可在固体内移动,而在900C时109个原子中则只有一个原子,试求其激活能(J/原子)。
10. 若将一块铁加热至850C,然后快速冷却到20C。
试计算处理前后空位数应增加多少倍(设铁中形成一摩尔空位所需要的能量为104600J)。
11. 设图1-18所示的立方晶体的滑移面ABCDF行于晶体的上、下底面。
若该滑移面上有一正方形位错环,如果位错环的各段分别与滑移面各边平行,其柏氏矢量b// AB1)有人认为“此位错环运动移出晶体后,滑移面上产生的滑移台阶应为4个b,试问这种看法是否正确?为什么?2)指出位错环上各段位错线的类型,并画出位错运动出晶体后,滑移方向及滑移量。
材料科学基础课后习题答案

(3) cosφ
=
n3 ⋅ F | n3 || F
|
=
1 3
cosα
=
b⋅F |b || F
|
=
1 2
由 Schmid 定律,作用在新生位错滑移面上滑移方向的分切应力为:
τ 0 = σ cosϕ cos λ = 17.2 ×
1× 3
1 = 7.0 MPa 2
∴作用在单位长度位错线上的力为:
f = τb = aτ 0 = 10 − 3 N/m 2
滑移面上相向运动以后,在相遇处
。
(B
)
A、相互抵消
B、形成一排空位
C、形成一排间隙原子
7、位错受力运动方向处处垂直与位错线,在运动过程中是可变的,
晶体作相对滑动的方向
。
(C
)
A、亦随位错线运动方向而改变 B、始终是柏氏矢量方向 C、始
终是外力方向
8、两平行螺型位错,当柏氏矢量同向时,其相互作用力
。
(B
二、(15 分)有一单晶铝棒,棒轴为[123],今沿棒轴方向拉伸,请分析:
(1)初始滑移系统; (2)双滑移系统 (3)开始双滑移时的切变量 γ; (4)滑移过程中的转动规律和转轴; (5)试棒的最终取向(假定试棒在达到稳定取向前不断裂)。
三、(10
分)如图所示,某晶体滑移面上有一柏氏矢量为
v b
的圆环形位错环,并受到一均匀
14、固态金属原子的扩散可沿体扩散与晶体缺陷扩散,其中最慢的扩
散通道是:
。
(A)
A、体扩散
B、晶界扩散
C、表面扩散
15、高温回复阶段,金属中亚结构发生变化时,
。
(C)
A、位错密度增大 B、位错发生塞积 C、刃型位错通过攀移和滑移构
材料科学基础章作业参考答案
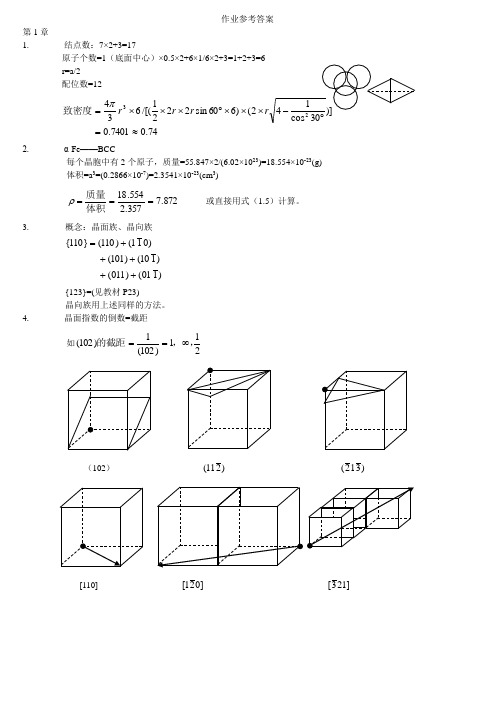
作业参考答案第1章1. 结点数:7×2+3=17原子个数=1(底面中心)×0.5×2+6×1/6×2+3=1+2+3=6r=a/2配位数=1274.07401.02()660sin2221/[(6343≈=⨯⨯⨯︒⨯⨯⨯=rrrπ致密度2. α-Fe——BCC每个晶胞中有2个原子,质量=55.847×2/(6.02×1023)=18.554×10-23(g)体积=a3=(0.2866×10-7)=2.3541×10-23(cm3)872.7357.2554.18===体积质量ρ或直接用式(1.5)计算。
3.概念:晶面族、晶向族)101()011()110()101()011()110(}110{+++++={123}=(见教材P23)晶向族用上述同样的方法。
4. 晶面指数的倒数=截距如211)102(1)102(,,的截距∞==(102))211()312( [110] ]021[]213[5.晶向指数:]101[和]011[6.7.8. 9. (略,不要求) 10.设晶格常数为a22100a =)面密度( 785.048210022==⨯=ππr r )面致密度( 222110a=)面密度( 555.02428211022==⨯=ππrr )面致密度(2234321111a r ==)面密度( 906.03232111122==⨯=ππr r )面致密度( 11. (略,不要求)12. (略,不要求) 13. 6/2+12/4=614.立方晶系晶面间距计算公式: 222lk h na d ++=① )nm (143.0286.02100121222100=⨯=++=ad)011()110()112(]011[]212[]111[)nm (202.0286.021011222110=⨯=++=a d)nm (0764.0286.0141321222123=⨯=++=a d②)nm (1825.0365.02100121222100=⨯=++=ad)nm (2107.0365.031111222111=⨯=++=a d)nm (09125.0365.042121121222112=⨯=++=ad③(略,不要求)15. (略,不要求) 16. (略,不要求)一、 单项选择题。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
计算材料学1.计算材料学在不同层次使用的计算方法2.材料科学与工程的四要素包括材料固有性质、结构与组分、使用性能、合成与加工。
3.根据晶体中原子间的结合力类型划分有哪几种晶体类型,其特点是什么?类型作用力键合特点离子键静电库仑引力最强无方向性、配位数高、熔点高、强度高、低膨胀系数、塑性差、导电性差、溶态导电共价键轨道相互作用强有方向性、配位数低、熔点高、强度高、低膨胀系数、塑性差、溶态不导电金属键原子实与自由电子之间的景点相互作用(库仑力)较强无方向性,配位数高,塑性好,导电性好,导热性好,结构密堆分子键瞬时偶极矩较弱无方向性,配位数高,结构密堆,熔点高,绝缘氢键氢原子核与极性分子间的库仑力弱无方向性,无饱和性当样品从绝对零度开始加热时,只有能量位于费米能附近kBT 范围内的轨道电子才被热激发,每个被激发电子所获得的能量量级正好为 kBT , 所以被激发电子的比例约为 T/TF ,设 N 为电子总数,则被激发的电子数约为 NT/TF ,在被激发的 NT/TF 个电子中,每个都具有能级为 kBT 的热能,所以总的电子热能的量级为,于是电子热容为,正比于温度 T ,与实验结果一致。
室温下, TF 约为 5×104 K ,Cel 比经典值 (3/2)NkB 约小两个量级。
5. 自由电子轮的两个假设(1) 假设自由电子在金属晶体中的恒定势场下运动(2) 单电子近似 6. 根据,T 的实验数据作图求出电子气的比热容常数()Tk T T NU FB ~7.自由电子近似与近自由电子近似的主要区别自由电子近似(遵循波尔兹曼统计规律):假设电子在金属晶体的恒定场中运动,在此模型下建立薛定谔方程;单电子近似,即自由电子理论把电子的运动状态从一个多体问题转化为单体问题处理,处理过程中考虑电子费米对称性。
近自由电子近似(遵循费米-狄拉克统计):除了自由电子近似的两个假设外,还大胆的做出如下近似(1)绝热近似:原子核(或离子实)的质量>>电子质量,离子运动速度慢,假设离子固定在瞬时位置上,多体问题化成多电子问题(2)自洽场(H-F)方法:多电子问题单电子问题,每个电子是在固定的离子势场及其它电子的平均场中运动(3)认为所有离子势场和其他电子的平均场是周期性势场。
8.能隙的起因9.二维晶体的第一、二、三布里渊区10. 二维正方点阵的第一布里渊区的等能面11. 说明为什么铜锌合金在50%为BCC 结构(计算β相下的电子密度)BCC 结构 Zn 含量大于35%,倒格子为FCC ,第一布里渊区为十四面体, 距离原点最近的位置=面心立方对角线14224aaππ=, 内切球的体积=333422)(2)33a aππππ= 设晶体有N 个院子,BCC 原胞体积为312a ,晶体体积Vc=332a L N =⋅。
在K 空间,3322()2(2)L Vcππ=,内切球内体积容纳的电子数=内切球体积×状态密度3322222(2)(2)Vc Vc N πππππ⋅==≈1.48N 12. 量子化学从头算的三个近似非相对论近似、Born-Oppenheimer 近似(绝热近似)、单电子近似(轨道近似)13. RHF 方程占据某空间轨道α电子与 β电子为同一空间坐标函数所描写,这就隐含了对波函数的一种限制,从这样的波函数推导得到的方程称为自旋限制的Roothaan 方程,简记为 RHF 方程。
其中,14. UHF 方程15. 自旋多重度包含 p 个α电子 和 q 个 β电子(p >q )的开壳层体系的自旋限制的单 Slater 行列式波函数16. 写出氢原子1s 轨道的波函数设外层贵大由2个STO 表示,其中1个STO 由3个GTO 展开的用Q1表示,由一个GTO 展开的用Q2表示,而总的波函数用Q 表示,则代入得231413.0071213.0070.0334946()3.14r Q e -⨯=⨯⨯2314 1.962082 1.962080.234727()3.14r e -⨯+⨯⨯23140.44452920.4445290.813757()3.14r e-⨯+⨯⨯ 223140.1219490.121949220.1219491.0()=0.347061293.14r r Q e e --⨯=⨯⨯⨯120.42743058Q +0.66544951Q Q =⨯⨯2213.007 1.96208=0.069911480.11858819r re e --⨯+⨯220.4445290.1219490.135008360.23095177r r e e --+⨯+⨯17. 密度泛函理论的基本思想18. 密度泛函理论的基本内容 1.Hohenberg-Kohn 定理①基态的总能可以写成是电荷密度的泛函 ②该总能的电荷密度泛函满足变分原理 2.Thomas-Fermi 模型推论 ②忽略交换-关联作用 ②增加交换-关联能 3.Kohn-Sham 泛函 将动能T [ρ]近似为T 0[ρ],求,求薛定谔方程,确定19. 证明:两个不同的外势不能给出相同的基态电子密度这是不可能的,所以两个不同的 V 不能给出相同的基态密度 ρ(r )。
20. 运动方程的形式经典力学方程、哈密顿方程,拉格朗日方程、牛顿力学方程 21. 常见的系综微正则系综(N 、V 、E 不变)——等能 正则系综(N 、V 、T 不变)——等温 等压系综(N 、P 、T 不变) 22. 计算机模拟方法的两个基本限制一是观测时间是有限的,二是有限的系综大小 23. 分子动力学模拟的基本步骤24. 势的模型,写出力的表达式U(r)()F r r ∂=-∂,126137U(r)1482r r r σσε⎡⎤∂=--⎢⎥∂⎣⎦,1261371()482F r r r σσε⎡⎤=-⎢⎥⎣⎦ 25. 写出初始化的方法26.写出趋衡的过程并讨论动能、势能及总能趋衡的特点执行趋衡过程的办法:对运动方程积分若干MD步(这里是50步),然后停止积分过程,对速度作专门的再调整,以取走或增加系统能量。
这种调整通过对全部粒子的速度重新标度来实现。
图中显示在数百步之后动能就达到平衡,在几百步后已给出确定的平均值。
而势能的弛豫要比前一组慢得多。
对速度每作一次重新标度,能量就跳跃到一个不同的值。
在两次速度标度之间,只要系统不受外部干扰,能量就保持几乎恒定。
能量并不保持严格恒定的原因,是我们没有使势能光滑地变为零。
这样,每当粒子进入到截断力程之内,就会引起能量的涨落。
27.根据计算结果(表4.1)讨论截断距离对势能、动能的影响对于一定的T*与ρ*,截断距离越大趋衡势能与动能的变化不是很大,差值也变化很小;对于相同的截断距离,不同的T*与ρ*趋衡势能与动能的变化很大,差值也变化很大。
原子间势的理解(表达式)原子相互靠近时会形成化学键,原子的结合是核和所有电子静电相互作用的结果,其相互作用可分为两类:吸引作用和排斥作用。
固体中的吸引作用全部归因于异性电荷之间的静电吸引相互作用。
排斥作用的来源一是同性电荷之间的库仑力,二是泡利原理引起的排斥作用。
28.表述对势存在的不足由于忽略了多体相互作用,对于包含非闭壳层原子的体系,这种两体近似是不适合的,而且所得结果与很多实验结果不一致。
对势模型的明显缺点是导致了实际金属中并不存在的Cauchy 关系C12 = C44 。
而且如果不考虑表面力的影响而接受Cauchy 关系的话,就会出现空位形成能与原子的内聚能相等的不合理的情况。
29.常见的几种对势Lennard-Jones势Morse势Born-Mayer势30.EAM势的基本思想EAM 势的基本思想是:某原子的原子核除了受到周围其他原子核的排斥作用外,还受到该原子核外电子及其周围其他原子产生的背景电子的静电作用。
即:把系统中的每个原子都看作是嵌入在由其他原子组成的基体中的客体原子,将系统的能量表示为嵌入能和相互作用能之和。
从而将多原子相互作用归结为嵌入能。
31.EAM势数学表达式及各项意义第一项为嵌入能,第二项为静电相互作用。
F 是吸引能,是所有其他原子在原子i 处产生的电子密度,φ是短程静电对势。
32. 常见的几种EAM 势的形式Daw-Baskes EAM Foiles-Daw-Baskes EAM Johnson-Oh EAM Mishin EAM33. 已知BCC 结构的金属Nb 的相互作用势1()2i ij i iE V r A ρ=-∑,其中()i ij jr ρφ=∑,2()(),()0,r r d r d r r d φφ=-≤=f ,22012()()(),()0,V r r c c c r c r r cV r r c =-++≤=f ,各参数如下:d=3.915354A o,c=4.20A o,c0=-1.5640104,c1=2.0055779,c2=-0.4663764,A=3.013798eV ,晶格常数a=3.3008 求i E近邻原子:1233,8,62,12r r a r a ===个个个,3r c f ,22012()()()V r r c c c r c r =-++23()8 3.3008 4.2)2V r =⨯-2331.5640104 2.0055779 3.30080.4663764( 3.3008)⎡⎤-+-⨯⎢⎥⎣⎦26(3.3008 4.2)+⨯-21.56401042.00557793.30080.4663764(3.3008)⎡⎤-+⨯-⨯⎣⎦=5.0323907662238(3.3008 3.915354)6(3.3008 3.915354)i ρ=⨯-+⨯-=11.20028661 i E =-7.570030927。