药物研发与技术审评沟通交流管理办法
CDE-76个常见一般性技术问题解答
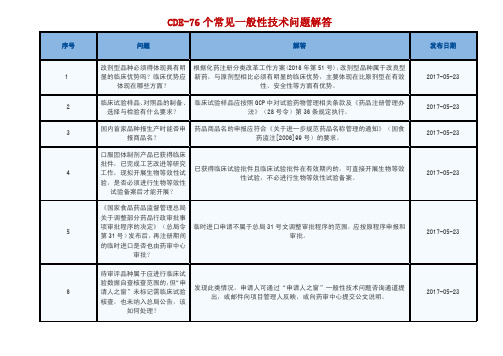
评,完成审评后由业务管理处按同审评序列注册申请的申报时间顺序制作 送签文件和放行。因此,申报时间靠后但先完成审评的注册申请,需等待 排在其前面的任务完成后才能转入下一环节。纳入优先审评程序的注册申
2017-05-23
请不受按序放行的限制,各环节均应第一时间处理。
国产注册申请应请向省局提出撤回申请,由省局把撤回申请和省局意见单
同规格品种” 项填写该制剂已被受理或关联审批的原料药、辅料、药包材、
关于原辅包关联申报的制剂申
23
请表填报问题
制剂或不同规格品种的受理号/登记号及名称。完成临床研究申请上市的申 请,还需填写原临床申请受理号、临床试验批件号、临床试验登记号或生
2018-03-12
物等效性试验备案号等。
按照《总局关于调整药品注册受理工作的公告》(2017 年第 134 号)规定:
关于根据药品批准文件要求完 按照《关于进一步规范药品注册受理工作的通知》(2015 年第 122 号)要
28
成上市后相关技术研究的申报 求,此类情形应按补充申请申报,按照《药品注册管理办法》附件 4 药品
2018-03-12
方式
补充申请注册事项第 18 项办理。
29Байду номын сангаас
关于已上市眼用制剂增加单剂 量装量规格的申报方式
相应平台进行备案。
33
关于进口产品多个包装厂的申 申请人可在申请表中机构 3(进口药品国外包装厂)一栏可填写多个包装厂
2018-03-12
序号
问题
解答
发布日期
报
名称和地址,各包装厂中间用分号间隔以示区别。
按照《药品注册管理办法》(局令第 28 号)第二十四条规定,申请人委托
药物研发与技术审评沟通交流管理办法

药物研发与技术审评沟通交流管理办法(试行)(修订稿)第一章总则第一条为规范申请人与国家食品药品监督管理总局药品审评中心(以下简称药审中心)之间的沟通交流,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)和《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)等有关规定,制定本办法。
第二条本办法所指的沟通交流,系指在药物研发过程中,经申请人提出,由药审中心项目管理人员(以下简称项目管理人员)与申请人指定的药品注册专员共同商议,并经药审中心适应症团队同意,就现行药物研发与评价指南不能涵盖的关键技术等问题所进行的沟通交流。
第三条沟通交流的形式包括:面对面会议、视频会议、电话会议或书面回复。
鼓励申请人与审评机构通过电话会议沟通。
沟通交流的提出、商议、进行,以及相关会议的准备、召开、记录和纪要等均应遵守本办法。
第四条本办法规定的沟通交流会议适用于创新药物、改良型新药、生物类似药、复杂仿制药以及一致性评价品种等研发过程和注册申请中的沟通交流。
第五条沟通交流是申请人与审评人员就技术问题互动的过程,双方在沟通交流过程中可就讨论问题充分阐述各自观点,最终形成的共识可作为研发和评价的重要依据。
第二章沟通交流会议类型第六条沟通交流会议分为Ⅰ类、Ⅱ类和Ⅲ类会议。
(一)Ⅰ类会议,系指为解决药物临床试验过程中遇到的重大安全性问题和突破性治疗药物研发过程中的重大技术问题而召开的会议。
(二)Ⅱ类会议,系指为创新药物在研发关键阶段而召开的会议,主要包括下列情形:1.临床试验申请前会议。
为解决首次递交临床试验申请前的重大技术问题,对包括但不限于下述问题进行讨论:现有研究数据是否支持拟开展的临床试验;临床试验受试者风险是否可控等。
对新机制新结构药物全球首次申报进行临床试验的,申请人应与审评机构进行沟通,明确申报技术要求。
2.Ⅱ期临床试验结束/Ⅲ期临床试验启动前会议。
为解决Ⅱ期临床试验结束后和关键的Ⅲ期临床试验开展之前的重大技术问题,对包括但不限于下述问题进行讨论:现有研究数据是否充分支持拟开展的Ⅲ期临床试验;对Ⅲ期临床试验方案等进行评估。
中药新药研究过程中沟通交流会议药学资料要求的指导原则

中药新药研究过程中沟通交流会议药学资料要求的指导原则(征求意见稿)2020年07月目录一、概述 (1)二、基本要求 (1)三、沟通交流会议药学资料要求 (2)(一)药物临床试验申请前会议 (2)1. 药物研究概况和药学研究资料 (2)2. 拟讨论问题 (3)(二)药物Ⅱ期临床试验结束/Ⅲ期临床试验启动前会议 (3)1.药物研究概况和药学研究资料 (4)2.拟讨论问题 (4)(三)药品上市许可申请前会议 (5)1.药物研究概况和药学研究资料 (5)2.拟讨论问题 (5)(四)其他会议 (6)四、参考文献 (6)五、著者 (7)中药新药研究过程中沟通交流会议药学资料要求的指导原则(征求意见稿)一、概述1沟通交流是申请人与国家药品审评机构解决研发问题2的有效方式,有利于解决中药新药研究及审评中存在的问题,3加速新药研发进程,促进中药传承创新。
为规范沟通交流会4议的药学资料,提高沟通交流的质量和效率,根据中药特点、5中药新药研发规律和沟通交流制度的相关规定,制定本指导6原则。
7本指导原则适用于中药新药研发过程中沟通交流会议8的药学资料,重点针对药物临床试验申请前、药物Ⅱ期临床9试验结束/Ⅲ期临床试验启动前、药品上市许可申请前的沟通10交流会议。
其他沟通交流会议的中药药学资料要求可参考。
11沟通交流会议的程序等参照国家药品审评机构相关会12议要求。
中药新药药学研究内容可参考相关指导原则和技术13要求。
14二、基本要求15坚持以问题为导向的基本原则,明确拟讨论问题。
申请16人应根据现行指导原则和技术要求等进行深入思考和充分17研究,围绕问题提供相关的背景信息、详实的研究资料和拟18解决方案,提供的资料同时应符合相关会议资料要求,以便19提高沟通交流的质量和效率,达到沟通交流会议的预期目的。
20申请人应基于不同研发阶段,提供相应的药学研究资料,21以便更好地评估已有研究数据是否支持拟开展的各期临床22试验、临床试验受试者安全风险是否可控、是否支持药品上23市许可的技术要求等。
国家药监局关于加强药品集中采购和使用试点期间药品监管工作的通知

国家药监局关于加强药品集中采购和使用试点期间药品监管工作的通知文章属性•【制定机关】国家药品监督管理局•【公布日期】2018.12.25•【文号】国药监药管〔2018〕57号•【施行日期】2018.12.25•【效力等级】部门规范性文件•【时效性】现行有效•【主题分类】药政管理正文国家药监局关于加强药品集中采购和使用试点期间药品监管工作的通知国药监药管〔2018〕57号各省、自治区、直辖市药品监督管理局,新疆生产建设兵团市场监督管理局:党中央、国务院高度重视人民健康福祉,为全面落实党中央、国务院重要决策部署和《国家组织药品集中采购试点方案》各项工作要求,切实保证药品集中采购和使用试点期间中标药品的质量,保障人民群众用药安全,现将有关要求通知如下:一、深刻认识试点工作重要意义国家组织药品集中采购和使用试点工作是党中央、国务院重要决策部署,各地要从政治和全局的高度,充分认识本次试点工作的重要性,切实把思想认识统一到党中央的决策部署上来,自觉增强“四个意识”,要以人民利益为中心,全力配合医改大局,全面落实药品监管“四个最严”要求,以监督检查和产品抽检为抓手,推动企业落实主体责任,切实保障药品质量安全。
二、加强药品生产监管各省级药品监管部门要坚持问题导向,强化日常监管,督促企业落实主体责任。
要加大对通过仿制药一致性评价品种特别是中标药品生产企业的现场检查力度,重点检查企业风险隐患排查责任落实情况、生产质量管理规范实施情况、数据真实可靠情况,严格落实原辅料质量控制,严控源头质量风险情况,严格按照批准的处方工艺组织生产情况,涉及委托生产的,落实委托生产质量管理情况。
要对照国家药品监管部门公告的通过仿制药一致性评价的品种建立台账,对通过日常监管发现应整改项目要逐项整改、逐一销账,确保通过仿制药一致性评价的品种质量安全。
要督促企业落实产品供应保障责任,严格执行药品停产报告工作要求,实事求是做好产能预估和各地投标工作。
新药 期临床试验申请技术指南

附件新药I期临床试验申请技术指南一、前言为帮助新药注册申请人(药品企业、科研机构和科研人员)申请I期临床试验,提高新药研发与审评效率,保护受试者安全与权益,保证临床试验质量,特发布本技术指南。
本指南阐述了新药在我国开展首次临床试验时需要向国家食品药品监督管理总局药品审评中心(以下简称药审中心)提供的信息。
本指南的目的是:明确新药I期临床试验的技术要求,提高I 期临床试验申报资料的质量;通过规范I期临床试验资料的数据要求,缩短新药研发周期,加快新药上市进程。
本指南适用于创新药和改良型新药,包括化学药品和治疗用生物制品(细胞和基因治疗产品除外)。
二、咨询与沟通交流如果申请人在研发及申请临床试验过程中有疑问,可通过药审中心网站查询相关指导原则,也可以按照相关规范通过药审中心网站“申请人之窗”就相关问题进行咨询。
递交新药临床试验申请前,申请人可按照《药物研发与技术审评沟通交流管理办法》所规定的方法与工作程序,申请与药审中心召开临床试验申请前会议。
申请人与药审中心间的沟通有助于提高临床试验申请的质量,加快后续研究与审评的进程。
三、I期临床试验申请的技术要求(一)资料格式及内容I期临床试验申请的申报资料应以纸质资料和电子资料方式提交,电子资料可以CD的形式送交。
格式和内容可参照研究人用药品注册技术要求国际协调会(ICH)通用技术文件(CTD)的要求整理提交。
(二)介绍性说明和总体研究计划介绍性说明应包括新药的名称、所有的活性成分、药理作用类别、结构式(如果已知)、剂型、制剂处方、给药途径、临床试验目的等。
如果有所研究药物用于临床的经验,应提供简短概述,包括在其他国家的研究和上市的经验;若没有,标题下写“无”。
总体研究计划应总结申请临床试验方案的设计依据,主要为拟定的适应症、受试者人群、受试者数量、给药方案(剂量、给药间隔、给药持续时间等)、药物安全性评价方法、风险控制计划等,根据已有信息预期的任何安全性(重要的已确定风险、重要的潜在风险、重要的缺失的资料等)的风险论证。
药物研发与技术审评沟通交流管理办法-EN

Administrative Regulation on the Communication for Drug R&D Activities andTechnical Review(Draft for Comment)Chapter I General ProvisionsArticle 1This regulation is formulated for the purpose of good performance of communication and standardization of the communication between the applicants and the Center for Drug Evaluation of NMPA (hereinafter referred to as CDE), by following the principles of the Pharmaceutical Administration Law of the People's Republic of China, and Measures for the Administration of Drug Registration.Article 2The communication, described in this regulation, is defined as the consultation and discussion applied by the applicants during the drug research and development process, through negotiation between the project management staff of CDE (hereinafter referred to as the “Project Manager”) and the drug registration specialist designated by the applicant, and approved by the review team of CDE, discussing the key technical issues that are not covered by the current drug R&D and review guidelines.Article 3The forms of communication include: face-to-facemeeting, video conference, teleconference and written correspondence. Applicants are encouraged to communicate with CDE through teleconference. The proposal, consultation and conduct of the communication, as well as the preparation, convening, recording and minutes of relevant meetings, shall comply with this regulation.Article 4The communication meetings stipulated in this regulation shall be applicable to the communication and consultation for the innovative drugs, modified new drugs, biosimilar drugs, Chinese herbal medicine compound preparation based on traditional classic prescription, drugs with the same name and prescription, complex generic drugs, and equivalency assessment drugs during their R&D process and registration applications.Article 5Communication is an interaction process dealing with the technical issues between the applicant and the review team, during which both parties may fully present their respective viewpoints with regard to such issues.Chapter II Types of Communication MeetingsArticle 6Communication Meetings are divided into Class I, Class II and Class III meetings, at which the communication takes place in respect to major issues at key stages.(I)Class I meeting is defined as the meeting held on thepurpose to address the major safety issues encountered during the clinical trials of drugs, and the major technical issues in the R&D process of the breakthrough therapeutic drugs, or held in other prescribed situations.(II)Class II meeting is defined as the meeting held at the key stages of drug R&D process, which mainly include the following situations:1.Pre-Clinical Trial Application meeting. To address majortechnical issues before the first submission of the clinical trial application, the following issues can be discussed, including but not limited to: whether the available research data supports the proposed clinical trial; whether the risk of clinical trial subjects is controllable. The communication meeting materials prepared by the applicant shall include complete information on the clinical trial protocols or drafts, existing pharmaceutical and non-clinical study data, and other research data. For the clinical trial applications of the first global applications of the drugs with new mechanism and new structure, the applicant shall initiate the communication with CDE, and the technical requirements on the registration shall be established clearly.2.End of Phase II clinical trial of drugs/Phase III pre-clinicaltrial meeting. To address major technical issues encounteredat the end of Phase II clinical trial and prior to the initiation of pivotal Phase III clinical trial, the following issues can be discussed, including but not limited to: whether the available research data can fully support the proposed Phase III clinical trial; make assessment on Phase III clinical trial protocol.3.Pre-meeting for drug marketing authorization. To explorewhether the available research data meets the technical requirements for the application for drug marketing authorization, the following issues can be discussed, including but not limited to: whether the available research data supports the technical requirements for the application for drug marketing authorization. The applicant for conditional approval and/or for priority review approval procedures shall communicate with the CDE before submitting the application for drug marketing authorization to NMPA.4.Risk assessment and control meeting. This meeting is tohave a discussion before NDA approval on the adequacy and controllability of the post-marketing risk control in order to assess and control the post-marketing risk.5.Other circumstances of Class II meetings as prescribed. (III)Class III meeting is defined as the meetings other than ClassI and Class II meetings.Article 7Applicants who have fallen into one of the following circumstances may propose the corresponding communication application based on the proposed studies or submissions in accordance with the provisions of the three classes of meetings mentioned above.(I)For clinical trial application of adding new indication, theapplicant shall carry out the research on the basis of available data in combination with the characteristics of the new indication, and make communication with CDE if necessary.(II)For the key technical issues found during the R&D process of drugs for urgent clinical needs or treatment of rare diseases, the applicant may make the communication application.(III)For the major R&D issues in the complex generic drugs and equivalency assessment or re-assessment drugs (e.g.selection of the reference preparation, evaluation criteria for bioequivalence, etc.), the applicant may make the communication application.(IV)For the protocol designed for the complicated important non-clinical studies (e.g. carcinogenicity studies), the applicant may make the communication application. (V)Where there is technical disputation in the review process after the applicant has received the inquiry consultation andsupplementary notification, the applicant may make the communication application if the applicant still has any difference regarding the comprehensive assessment results. (VI)Application for communications can be made in the R&D process of drugs on the frontier technology field.Chapter III Proposal and Discussion of CommunicationMeetingsArticle 8The convening of a communication meeting shall meet the following basic conditions:(I)The “Application Form for Communication Meeting”(Annex 1) and “Communication Meeting Materials”(Annex 2) submitted shall comply with the requirements of the regulation;(II)"Communication Meetings Materials” and the "Application Form for Communication Meetings" shall be submitted at the same time;(III)The professional background of personnel participating in the communication meeting shall be qualified for the discussion on professional issues.Article 9Where the above communication requirements are met, the applicant shall submit the Application Form for Communication Meeting and Communication MeetingsMaterials through the “window of applicant” of the website of CDE, specifying the type of communication at application. Article 10Within 2 days of receiving the “Application Form for Communication Meeting”, the Project Manager shall complete the preliminary review according to requirements above. Where the materials are found not complete or to have fallen into other cases of nonconformity, the application shall be ended. Those meeting the requirements shall be further submitted to relevant professional review team.Within 18 days after the application, the review team shall determine how to carry out the communication: communication meeting (face-to-face meeting, video conference, teleconference) or written feedback. If the review team finds that any data is incomplete or inconsistent, the application shall be terminated directly.Article 11Within 20 days after the application, the Project Manager will notify the applicant the conclusion confirmed by the review team.For the communication meeting application approved, the Project Manager will notify the meeting agenda to the applicant through the “window of applicant” within 5 days after the confirmation of meeting date, including the date, place, matters needing attention and materials to be further discussed at the meeting.Article 12Communication meeting application may not beapproved for the following circumstances:(I)Additional data shall be provided before satisfying thecommunication requirements for the issues to be discussed; (II)The professional background of attendees is not qualified for the discussion of the technical issues and not satisfactory to the need for communication;(III)Other circumstances that do not guarantee the effective convening of the meeting.If the communication meeting cannot be convened, the Project Manager shall provide the specific reasons through the “window of applicant”. After finishing relevant works, the applicant can make a new communication application.Article 13If the communication meeting is approved, in general, Class I meeting will be held within 30 days after the application, Class II meeting will be held within 60 days after the application, and Class III meeting, within 75 days after the application.If the communication meeting is confirmed not to be held, the feedback shall be completed within the above-mentioned time limit according to the meeting type, in principle.Chapter IV Preparation for Communication MeetingsArticle 14The applicant shall submit the electronic version ofCommunication Meeting Materials in accordance with the requirements of the “Communication Meeting Materials” through the "Window of Applicant".Article 15To ensure the quality and efficiency of communication meetings, the drug registration specialist shall make comprehensive discussion with Project Manager before the meeting, and confirm the date, time, place and agenda of the meeting.Chapter V Convening of Communication MeetingsArticle 16The communication meeting will be chaired by the CDE staff, and conducted according to the pre-determined meeting agenda. Discussion shall be made on proposed issues put forward before the meeting. In principle, it is not within the scope of communication to raise new meeting materials, generate divergent issues and to temporarily add new issues. In general, the communication meeting duration shall be within 45 minutes, but not exceed 60 minutes.Article 17The minutes of meeting shall be prepared in accordance with the "Template of Communication Meeting Minutes" (Annex 3). Where both parties have reached an agreement, viewpoints in common shall be specified. If the parties fail to reach an agreement, their viewpoints shall be statedrespectively. The applicant is encouraged to make minutes of meeting on the spot, for the purpose of which the applicant can make a draft of the minutes before the meeting on the basis of the preliminary feedback from CDE and make it clear after discussion. If the minutes cannot be made on the spot for special reasons, the applicant shall generally submit the draft of minutes within one week after the meeting is completed. Minutes of meeting shall be finalized within 30 days upon the completion of the meeting. The Project Manager shall upload the minutes within 2 days upon finalization, to the communication system which can be accessed by the applicant through the “Window of Applicant”. Meeting minutes shall be archived as an important document. Article 18If necessary, CDE will make audio or video recordings of whole procedure of the meeting, and archive them as working files for future reference. The applicant and other attendees are not allowed to make any audio, video or photo recordings without permission. Where the applicant’s trade secrets are involved, CDE shall keep such trade secrets confidential in accordance with applicable laws.Chapter VI Postponement or Cancellation ofCommunication MeetingsArticle 19The meeting will be postponed if any of the following circumstances occurs:(I)The communication conditions are met but additionalinformation shall be supplemented;(II)The review team considers that there are other important issues beyond the meeting topic that need further discussion; (III)There are too many meeting materials for the review team to review in a limited period;(IV)The important attendees cannot attend the meeting on time; (V)Other force majeure factors.In general, the applicant shall be notified of the decision to postpone a meeting, at least 5 days prior to the meeting. The postponed meeting will be discussed by the Project Manager and the drug registration specialist. In general, the delay time shall not exceed 2 months. It will be considered that the meeting cannot be held if the delay time is more than 2 months due to the applicant, in which case the applicant is required to submit a new communication application after supplementing additional materials.Article 20 A meeting will be cancelled if any of the following situations occurs:(I)The meeting materials are not submitted within thespecified date;(II)The submitted meeting materials do not meet the requirements of this regulation;(III)The applicant makes the proposal of canceling the meeting which is approved by CDE;(IV)The issue from the applicant has been resolved or has been given written correspondence.In general, the decision to cancel a meeting shall be informed to the applicant 5 days prior to the meeting. After cancellation of the meeting, the applicant may submit a separate communication application according to this regulation.Chapter VII Supplementary ProvisionsArticle 21For the verification or consultation of general technical issues, the applicant may communicate with the Project Manager through the general technical issue consultation platform in "Window of Applicant", telephone, fax, E-mail, or by other means. Consultations on general technical issues will not include the key technical issues in drug R&D and technical review processes.Article 22When making submission of the drug registration dossier, the applicant shall appoint 1 or 2 drug registrationspecialists, and provide the specific information of the drug registration specialist such as the name and telephone number. The drug registration specialist shall be responsible for drug registration. The applicant shall make communication with CDE through the drug registration specialist, and the Project Manager shall only contact the drug registration specialist designated by the applicant. If there is any change of the drug registration specialist, the applicant shall make updates through the “Window of Applicant” in a timely manner.Article 23During the review process, CDE may make a proposal on communication as required, which will be discussed by Project Manager with the drug registration specialist to determine the, time, agenda, and information requirements for the communication meeting.Article 24The meeting materials used for communication shall be included in the applied registration documents (which may be compiled into an individual volume) as the basis for review. Important meeting materials prior to the submission of a drug registration application, e.g. Communication Meetings Materials, Application Form for Communication Meeting and meeting minutes, should be submitted by incorporating into the dossiers. The meeting materials generated during the review should be included into the dossiers by CDE.Article 25CDE staff shall strictly follow this regulation, andmust not contact the applicant privately in any way other than those provided in this regulation. The special case shall be approved by CDE.Article 26This regulation shall come into force as of the date of promulgation.Annexes:1.Application Form for Communication Meetingmunication Meeting Materials3.Template of Communication Meeting MinutesAnnex 1Application Form for Communication MeetingI.Basic information of drug research and development1.Applicant2.Drug name3.Acceptance number (if available)4.Chemical name and structure (or prescription, for ChineseTraditional Medicine)5.Proposed indications (or major functions)6.Dosage form, administration route and regimen (medicationfrequency and treatment cycle)7.Drug R&D strategies, including drug R&D backgroundinformation, drug development plans, brief description of the R&D process, key events, and current development status. II.Specific contents of meeting application1.Meeting types: Class I, Class II, or Class III.2.Classification of meeting: such as Pre-Clinical trialapplication meeting, End of Phase II clinical trial/Phase III pre-clinical trial meeting, Pre-meeting for drug marketing authorization or risk evaluation and control meeting.3.Meeting forms: face-to-face meeting, video conference,teleconference or written correspondences.4.Meeting objective: brief description.5.Proposed meeting date and time: please provide 3 alternatives.6.Proposed meeting agenda: including estimated duration foreach topic to be discussed (in general, the discussion duration of all topics should be controlled within 60 minutes.)7.List of attendees of applicant: the list shall set forth the namesof attendees, including the positions, job contents and work units. If the applicant plans to invite experts and interpreters to attend the meeting, they shall also be involved in the list.8.List of proposed issues under discussion: it is recommendedthat the applicant shall make the list of issues by discipline, including but not limited to the issues of the pharmaceutical, pharmacology & toxicology, and clinical practice. Each issue should include a brief description of background, the purpose of proposed issues and the applicant’s opinions of issues. In general, the number of issues to be discussed at a meeting shall not exceed 10.Annex 2Communication Meeting Materials1.List of issues under discussion: a list of issues that applicanthas finalized. It is recommended that the applicant shall make the list of issues by discipline, including but not limited to the issues of the pharmaceutical, pharmacology & toxicology, and clinical practice. Each issue shall include a brief description of background and the purpose of issues proposed.2.Supporting data summary: summary of supporting data bydiscipline and the order of issues.In terms of the summary of supporting data, relevant studies, results and conclusions shall be explained by data. Taking end of Phase II clinical trial meeting as an example, the clinical professional summary shall include the following: (1) a brief summary of completed clinical trials, including data, results and conclusions. Important dose-effect relationship information shall also be included. In general, a complete clinical study report does not need to be provided; (2) a detailed introduction of the proposed Phase III clinical trial protocol to confirm the main characteristics of clinical trials, such as clinical trial subject population, the key inclusion and exclusion criteria, clinical trialdesign (e.g., randomization, blinding, control selection, justification of non-inferiority threshold if non-inferiority trial used), dose selection, primary and secondary efficacy endpoints, and primary analysis methods (including planned interim analysis, characteristics of adaptation studies, and major safety concerns).Annex 3Template of Communication Meeting MinutesMeeting types: Class I, Class II, or Class III.Classification of meeting: such as Pre-clinical trial application meeting, End of Phase II clinical trial/Phase III pre-clinical trial meeting, Pre-meeting for drug marketing authorization or risk evaluation and control meeting.Date and time of the meeting:Meeting place:Acceptance number (if available):Drug Name:Proposed indications (or major functions):Applicant:Chairman:Recorder:Attendees: a list of all attendees of the applicant and CDE.Text:1.Purpose of the meeting:2.Meeting background:3.Issues discussed at the meeting and results:(1)Issue 1: XXXXXXXXXDo both sides reach consensus?□ Yes.Common opinion: XXXXXXXXX □ No.Applicant's comments:CDE's comments:(2)Issue 2: XXXXXXXXX…。
CDE沟通交流
二、药品注册申请中沟通交流
b、对于中心正式的书面交流存在问题的: 即是:补充资料通知中的问题 或批件中要求研究的问题 • 沟通方式:信息反馈、电话咨询、咨询日 • 需要的准备:准确理解问题;人员专业对 口
三、特殊审批程序
第2、3个内容主要是针对特殊审批程序 的,目前我们单位没有此类项目,大概了 解下这类申请,我们和CDE的沟通,可以 有个早期沟通交流申请。2010年CDE共收 到早期沟通交流申请35个,同意召开的早 期特殊审批沟通交流会议15个,其他未获 批准的主要原因:问题不明确、依据不充 分、人员结构不合理。
四、企业、CDE沟通交流体会
• CDE的角度希望我们准备问题时要考虑: a、避免过于开放的问题; b、准确定位; c、阐述问题产生的背景和数据; d、提出解决问题的建议和想法; e、存在的顾虑和疑虑。
四、企业、CDE沟通交流体会
• 另要避免一些误区: a、避免寻找答案的交流; b、避免推诿责任的交流; c、避免只问不答,不提供依据的交流; d、避免没有处理建议的交流; e、避免没有积极反馈的交流; f、 避免责怪似的交流。
圆桌会议视频会议电话会议等五关于审评会议中的沟通交流企业沟通会的三种类型cde根据审评需要主动提出需与申报单位进行沟通交流凡符合特殊审评程序的品种其申报单位可根据相关管理规定提出与cde进行沟通交流因其他情形需要会议沟通时五关于审评会议中的沟通交流详细的会前准备应对突发事件的预案五关于审评会议中的沟通交流复审三方审议会复审
八、CDE网站信息公开与交流
• 电子提交范围(2011年度提交要求) 1、申请生产的品种需电子提交的内容为:
(1)质量标准 1 (6)立题目的与依据 6 (2)药品使用说明书(7)对主要研究结果的总结与评价 (3)包装标签 (8)药学研究资料综述 (4)工艺资料 (9)药理毒理研究资料综述 (5)起草/修订说明(10)临床试验研究资料综述
审评审批解读
药物临床试验审评审批流程
沟通交 流会召
开
开展临 床试验
01
02
03
04
沟通交 流申请
IND申 请
01 沟通交流会议的准备与申请
• 申请人准备的沟通交流会议资料应包括:临床试验方案或草案、对已有的 药学和非临床研究数据及其他研究数据的完整总结资料。
02 沟通交流会议的召开
会议由药审中心工作人员主持,双方围绕药物临床试验方案就申请人提 出的关键技术问题,以及已有资料和数据是否支持实施临床试验开展和受 试者安全风险是否可控进行讨论,并为后续研究提出要求和建议。
沟通交流会议应按《沟通交流办法》要求形成会议纪要。会议纪要作为 审评文档存档,并作为审评和审批的参考。
• 申请人应自行评估现有的研究是否符合申报拟实施临床试验的基本条件, 并明确拟与药审中心讨论的问题。
• 提交药审沟通交流会议申请表,中心对沟通交流会议资料进行初步审评
会议类型
会议内容
Ⅰ类会议
Ⅱ类会议 I期临床试验 申请前会议
指为解决药物临床试验过程中遇到的重大安全性问题和突破性治疗药物研发 过程中的重大技术问题而召开的会议。
风险评估和 控制会议
Ⅲ类会议
为评估和控制药品上市后风险,在批准新药上市前,对药品上市后风险控制 是否充分和可控进行讨论。
指对创新药物除Ⅰ类和Ⅱ类会议之外的其他会议,还包括一般改良型新药和 仿制药研发过程中遇到的重大问题而召开的会议
会议资料提交时间:Ⅰ类会议的《沟通交流会议资料》应与 《沟通交流会议基本信息表》同时提交,Ⅱ类和Ⅲ类会议的 《沟通交流会议资料》应在会议召开30日前提交。
药物临床试验审评审批流程解读
180724关于调整药物临床试验审评审批程序的公告(2018年第50号)
关于调整药物临床试验审评审批程序的公告(2018年第50号)颁布时间:20180724为鼓励创新,加快新药创制,满足公众用药需求,落实申请人研发主体责任,依据中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),对药物临床试验审评审批的有关事项作出调整:在我国申报药物临床试验的,自申请受理并缴费之日起60日内,申请人未收到国家食品药品监督管理总局药品审评中心(以下简称药审中心)否定或质疑意见的,可按照提交的方案开展药物临床试验。
现就具体事宜公告如下:一、沟通交流会议的准备与申请(一)申请人在提出新药首次药物临床试验申请之前,应向药审中心提出沟通交流会议申请,并在确保受试者安全的基础上,确定临床试验申请资料的完整性、实施临床试验的可行性。
(二)申请人准备的沟通交流会议资料应包括临床试验方案或草案、对已有的药学和非临床研究数据及其他研究数据的完整总结资料。
申请人应自行评估现有的研究是否符合申报拟实施临床试验的基本条件,并明确拟与药审中心讨论的问题。
(三)申请人应按照《药物研发与技术审评沟通管理办法(试行)》(以下简称《沟通交流办法》)要求,提交沟通交流会议申请表(附件1)。
药审中心应及时通知申请人是否召开沟通交流会议,并与申请人商议会议时间。
申请人应按沟通交流相关要求按时提交完整的沟通交流会议资料(附件2)。
药审中心对沟通交流会议资料进行初步审评,在沟通交流会议召开至少2日前,通过“申请人之窗”将初步审评意见和对申请人所提出问题的解答意见告知申请人。
申请人在收到初步审评意见和解答意见后,应尽快反馈问题是否已经得到解决。
申请人认为问题已经解决不需要召开沟通交流会议的,应通过药审中心网站“申请人之窗”告知药审中心取消沟通交流会议申请;申请人认为申请沟通交流的问题仍未得到解决的,按原定计划继续组织会议召开。
二、沟通交流会议的召开(四)会议由药审中心工作人员主持,双方围绕药物临床试验方案就申请人提出的关键技术问题,以及已有资料和数据是否支持实施临床试验开展和受试者安全风险是否可控进行讨论,并为后续研究提出要求和建议。
国家食品药品监督管理局药品审评中心加强对外沟通交流 促进审评公开透明
次 。咨询会议审议 品种 3 2 ( 9 个 创新药物 6 个 ,其他 药物 3 5 ) 7 2 个 ,邀请审评专家 3 8 人次 ,注册 申请人代表 2 4 26 0 0人次。
三 方共 同就 技 术 审评 中 的问 题进 行 深 入 的 沟 通 交 流 。另 在 过 渡 期 品 种 集 中审 评 工 作 中 ,药 品审 评 中 心 组 织 召 开 院 士 论 证
中国药事 2 0 0 9年第 2 3卷第 3 期
27 5
所谓黄金线是指 对于男性顾 客而言 货架上 8 5 4
1 5m 的高度 范 围 ,女 性 顾 客 7 ~ 1 5m 的 高 度 3c 5 2c 范 围 ;而 次要 高 度对 于男 性 顾 客是 7 ~ 8 c 或 5 5m
1 5 1 5m,女 性 顾 客 是 6 3 ~ 4c 0~ 7 c 或 15~ 5m 2
第2 6号 ,20 . 07
l 左 右内陈列 的商品 。 m 实践也证 明 .两 种陈列所带来 的效果 是不一样 的。纵 向陈列 能使 系列 商 品体 现 出直 线式 的效果 , 使 顾客一 目了然 ,系 列商 品纵 向 陈列 会 使 2 ~ 0
8 的商 品销量增加 。 O
[ ] 处方药与非处方药分类管理办法 ( 4 试行)E 3.国家药品监 s
会 、专家论证会、大型企业代表 座谈会 、中化药技术 审评技 术要求讨 论会 、品种专
20 人 次 ,涉 及 品种 近 万 个 。 00
二 、召开与注册 申请人沟通交流会议 。2 0 ,药品审评中心召开 与申请 人的沟通交流会议 3 0 8年 6次 ,与 2 0余名注册 0
集 店长 、员 工及顾客 的意见 和建 议 ,及 时调整 ,这
样 才能发挥 出培训 的作 用 。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药物研发与技术审评沟通交流管理办法(试行)(修订稿)第一章总则第一条为规范申请人与国家食品药品监督管理总局药品审评中心(以下简称药审中心)之间的沟通交流,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)和《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)等有关规定,制定本办法。
第二条本办法所指的沟通交流,系指在药物研发过程中,经申请人提出,由药审中心项目管理人员(以下简称项目管理人员)与申请人指定的药品注册专员共同商议,并经药审中心适应症团队同意,就现行药物研发与评价指南不能涵盖的关键技术等问题所进行的沟通交流。
第三条沟通交流的形式包括:面对面会议、视频会议、电话会议或书面回复。
鼓励申请人与审评机构通过电话会议沟通。
沟通交流的提出、商议、进行,以及相关会议的准备、召开、记录和纪要等均应遵守本办法。
第四条本办法规定的沟通交流会议适用于创新药物、改良型新药、生物类似药、复杂仿制药以及一致性评价品种等研发过程和注册申请中的沟通交流。
第五条沟通交流是申请人与审评人员就技术问题互动的过程,双方在沟通交流过程中可就讨论问题充分阐述各自观点,最终形成的共识可作为研发和评价的重要依据。
第二章沟通交流会议类型第六条沟通交流会议分为Ⅰ类、Ⅱ类和Ⅲ类会议。
(一)Ⅰ类会议,系指为解决药物临床试验过程中遇到的重大安全性问题和突破性治疗药物研发过程中的重大技术问题而召开的会议。
(二)Ⅱ类会议,系指为创新药物在研发关键阶段而召开的会议,主要包括下列情形:1.临床试验申请前会议。
为解决首次递交临床试验申请前的重大技术问题,对包括但不限于下述问题进行讨论:现有研究数据是否支持拟开展的临床试验;临床试验受试者风险是否可控等。
对新机制新结构药物全球首次申报进行临床试验的,申请人应与审评机构进行沟通,明确申报技术要求。
2.Ⅱ期临床试验结束/Ⅲ期临床试验启动前会议。
为解决Ⅱ期临床试验结束后和关键的Ⅲ期临床试验开展之前的重大技术问题,对包括但不限于下述问题进行讨论:现有研究数据是否充分支持拟开展的Ⅲ期临床试验;对Ⅲ期临床试验方案等进行评估。
3.提交新药上市申请前会议。
为探讨现有研究数据是否满足新药上市审查所需资料要求,对包括但不限于下述问题进行讨论:现有研究数据是否支持新药上市申请审查所需资料要求。
经讨论符合要求,或经补充完善后符合要求的,方可向国家食品药品监督管理总局递交新药上市申请。
4.风险评估和控制会议。
为评估和控制药品上市后风险,在批准新药上市前,对药品上市后风险控制是否充分和可控进行讨论。
(三)Ⅲ类会议,系指对创新药物召开的除Ⅰ类和Ⅱ类会议之外的其他会议。
第七条申请人有以下情形的,可按照Ⅰ类、Ⅱ类、Ⅲ类会议规定提出沟通交流。
(一)拟增加新适应症的临床试验申请,申请人应结合新适应症特点,在已有数据基础上开展相应的研究,必要时可与药审中心沟通交流。
(二)临床急需或治疗罕见病的药物研发过程中的关键技术问题,申请人可提出沟通交流申请。
(三)一致性评价品种的重大研发问题(参比制剂的选择、生物等效性的评价标准等),申请人可提出沟通交流申请。
(四)复杂或无明确指导原则的重要非临床研究(致癌性研究,扩展的发育毒性研究等)的设计方案,申请人可提出沟通交流申请。
(五)审评过程中有技术分歧的,申请人可提出沟通交流申请。
(六)对前沿技术领域药物研发过程中的沟通交流申请。
药审中心需追踪前沿技术进展、制定相应研发技术指南的,应成立专门工作小组与申请人在研发和评价过程中保持沟通交流。
第三章沟通交流会议的提出与商议第八条召开沟通交流会议应符合以下基本条件:1.提交的《沟通交流会议申请表》(附件1.1)和《沟通交流会议资料》(附件1.2)应满足本办法要求;2.《沟通交流会议资料》应在本办法规定时间内提交:Ⅰ类会议的《沟通交流会议资料》应与《沟通交流会议申请表》同时提交,Ⅱ类和Ⅲ类会议的《沟通交流会议资料》应在会议召开30日前提交;3.参加沟通交流会议人员的专业背景,应当满足针对专业问题讨论的需要。
第九条符合上述沟通交流条件的,申请人应通过药审中心网站“申请人之窗”提交《沟通交流会议申请表》,申请时应注明面对面会议、视频会议、电话会议或仅书面回复。
第十条项目管理人员收到《沟通交流会议申请表》后,应在3日内送达适应症团队组长,适应症团队组长与团队成员讨论后应在申请后10日内通过项目管理人员是否同意沟通交流。
第十一条确定召开沟通交流会议的,项目管理人员需在确定后5日内通过“申请人之窗”将会议议程告知药品注册专员,包括会议类型、日期、地点、会议内容,以及药审中心拟参会人员等信息。
第十二条有以下情形的,不能召开沟通交流会议:(一)拟沟通交流的问题,还需要提供额外数据才具备沟通交流条件的;(二)参会人员专业背景,不能满足沟通交流需要,无法就技术问题进行沟通的;(三)没有在本办法规定的时间内提交《沟通交流会议资料》的;(四)不能保证有效召开会议的其他情形。
不能召开沟通交流会议的,项目管理人员应当向药品注册专员说明具体原因。
申请人需在完善相关工作后,另行提出沟通交流。
第十三条确定召开沟通交流会议的,Ⅰ类会议一般安排在提出沟通交流后30日内召开,Ⅱ类会议一般安排在提出沟通交流后60日内召开,Ⅲ类会议一般安排在提出沟通交流后75日内召开。
第四章沟通交流会议的准备第十四条申请人应按照《沟通交流会议资料》要求提交电子版沟通交流会议资料。
电子资料通过“申请人之窗”中的“沟通交流”平台提交。
第十五条为保证沟通交流会议质量和效率,会议前药品注册专员应与项目管理人员进行充分协商。
药审中心参会人员应在沟通交流会议前对会议资料进行全面审评,并形成初步审评意见。
第十六条在正式会议召开至少2日前,项目管理人员通过“申请人之窗”将初步审评意见告知申请人。
在正式会议前申请人不应提出新问题。
若申请人认为问题已经得到解决的,应通过“申请人之窗”告知项目管理人员撤消沟通交流申请。
项目管理人员确认后应及时通知药审中心相关参会人员,将审评意见作为双方共识存档,同时记录取消会议的原因。
申请人认为大部分问题已经解决的,可申请将面对面会议改为电话会议等其他形式讨论。
第五章沟通交流会议的召开第十七条沟通交流会议由药审中心工作人员主持,依事先确定的会议议程进行。
项目管理人员全程参与会议,并记录会议情况。
一般情况下,沟通交流会议时间为60分钟。
第十八条会议纪要应按照《沟通交流会议纪要模板》(附1.3)要求撰写,经适应症团队审核确认,由项目管理人员上传至沟通交流系统,申请人可通过申请人之窗查阅。
会议纪要主要包括会议共识和会议分歧两部分内容。
鼓励当场形成会议纪要,会议纪要作为重要文档存档,并作为药物研发、审评和审批的重要依据。
第十九条药审中心必要时对会议进行全程录音、录像,作为工作档案存档备查。
申请人及其他参会人员未经许可,不得擅自录音、录像。
第六章沟通交流会议的延期或取消第二十条存在下列情形之一的,会议延期:(一)会议资料不充分,需要补充更多信息的;(二)申请人在会议议题之外增加了其他拟讨论的问题,或药审中心认为会议议题之外有其他重要问题需要进一步讨论的;(三)会议资料过多,以至于药审中心没有足够时间审评的;(四)关键参会人员无法按时参会的;(五)其他不可抗力因素等。
会议延期由项目管理人员与药品注册专员商议,一般延期时间不应超过2个月,超过2个月的,视为不能召开会议,申请人需完善资料后,另行提出沟通交流。
第二十一条存在下列情形之一的,会议取消:(一)会议资料没有在确定的日期内提交的;(二)提交的会议资料不符合本办法要求的;(三)申请人提出取消会议并经药审中心同意的。
会议取消后,申请人可按本办法要求另行提出沟通交流。
第七章附则第二十条申请人需要对一般性技术问题进行核实或咨询时,可以通过“申请人之窗”一般性技术问题咨询平台、电话、传真、邮件等形式与项目管理人员进行沟通交流。
一般性技术问题的咨询,不对药物研发与技术审评过程中关键性技术问题进行讨论。
第二十三条申请人在提交药品注册申请时,应指定1—2名药品注册专员,并提供药品注册专员的姓名、电话等具体信息和联系方式,药品注册专员专门负责药品注册事宜。
申请人应通过药品注册专员与药审中心进行沟通,项目管理人员也仅与申请人指定的药品注册专员进行接洽。
如果药品注册专员发生变更,申请人应及时书面告知药审中心。
第二十四条药审中心在审评过程中根据需要提出沟通交流由项目管理人员与药品注册专员商议,确定沟通交流会议时间、议程和资料要求。
第二十五条用于沟通交流的会议资料,应归入申报资料(可单独整理成卷)作为审评依据。
第二十六条药审中心工作人员应严格执行本办法,不得通过本办法规定之外的其他方式与申请人私下接触,特殊情况需经药审中心批准。
第二十七条本办法自发布之日起施行。
附件:1.1.沟通交流会议基本信息表1.2.沟通交流会议资料1.3.沟通交流会议纪要模板附件1.1沟通交流会议申请表一、药物研发基本情况1.申请人2.药品名称3.受理号(如适用)4.化学名称和结构(中药为处方)5.拟定适应症(或功能主治)6.剂型、给药途径和给药方法(用药频率和疗程)7.药物研发策略,包括药物研发背景资料、药物研制计划、研发过程的简要描述和关键事件、目前研发状态等。
二、会议申请具体内容1.会议类型:Ⅰ类、Ⅱ类或Ⅲ类。
2.会议分类:如I期临床试验申请前会议、Ⅱ期临床试验结束/Ⅲ期临床试验启动前会议,或提交新药上市申请前会议等。
3.会议形式:面对面会议、视频会议、电话会议或书面回复。
4.会议目的:简要说明。
5.建议会议日期和时间:请提供3个备选时间。
6.建议会议议程:包括每个议题预计讨论的时间(一般情况下,所有议题讨论时间应控制在60分钟以内)。
7.申请人参会名单:列出参会人员名单,包括职务、工作内容和工作单位。
如果申请人拟邀请专家和翻译参会,应一并列出。
8.建议参会适应症团队:如消化适应症。
9.提交会议资料时间:对于Ⅰ类会议申请,应同时提交《沟通交流会议资料》;对于Ⅱ类和Ⅲ类会议申请,提交《沟通交流会议资料》的时间不应少于会议前30天。
10.拟讨论问题清单:建议申请人按学科进行分类,包括但不限于从药学、药理毒理学和临床试验方案的设计等方面提出问题,每个问题应该包括简短的研发背景解释,该问题提出的目的及申请人对该问题的意见。
一般情况下,一次会议拟讨论的问题不应超过10-15个。
附件1.2沟通交流会议资料一、药物研发基本情况1.申请人2.药品名称3.受理号(如适用)4.化学名称和结构(中药为处方)5.拟定适应症(或功能主治)6.剂型、给药途径和给药方法(用药频率和疗程)7.药物研发策略,包括药物研发背景资料、药物研发计划、研发过程的简要描述和关键事件、目前研发状态等。