美国环保局 EPA 试验 方法9012bTotal and Amenable Cyanide (Automated Colorimetric, with Off-Line Dis
美国环保局 EPA 试验 方法 550_1

Analyte Acenaphthene Acenaphthylene Anthracene Benzo(a)anthracene Benzo(a)pyrene Benzo(b)fluoranthene Benzo(g,h,i)perylene Benzo(k)fluoranthene Chrysene Dibenzo(a,h)anthracene Fluoranthene Fluorene Indeno(1,2,3-cd)pyrene Naphthalene Phenanthrene Pyrene 1.2
550.1-1
METHOD 550.1 DETERMINATION OF POLYCYCLIC AROMATIC HYDROCARBONS IN DRINKING WATER BY LIQUID-SOLID EXTRACTION AND HPLC WITH COUPLED ULTRAVIOLET AND FLUORESCENCE DETECTION
Hale Waihona Puke July 1990J.W. Hodgeson W.J. Bashe (Technology Applications Inc.) T.V. Baker (Technology Applications Inc.)
ENVIRONMENTAL MONITORING SYSTEMS LABORATORY OFFICE OF RESEARCH AND DEVELOPMENT U.S. ENVIRONMENTAL PROTECTION AGENCY CINCINNATI, OHIO 45268
2.0
SUMMARY OF METHOD 2.1 Polycyclic aromatic hydrocarbons and internal standards, if used, are extracted from a water sample by passing 1 L of sample through a cartridge containing about 1 g of a solid inorganic matrix coated with a chemically bonded C-18 organic phase (liquid-solid extraction, LSE). The use of disks impregnated 550.1-2
美国环保局 EPA 试验 方法 613

APPENDIX A TO PART 136METHODS FOR ORGANIC CHEMICAL ANALYSIS OF MUNICIPAL ANDINDUSTRIAL WASTEWATERMETHOD 613—2,3,7,8-TETRACHLORODIBENZO-P-DIOXIN1.Scope and Application1.1This method covers the determination of 2,3,7,8-tetrachlorodibenzo-p-dioxin(2,3,7,8-TCDD). The following parameter may be determined by this method:Parameter STORET No.CAS No.2,3,7,8-TCDD....................................346751746-01-6 1.2This is a gas chromatographic/mass spectrometer (GC/MS) method applicable to thedetermination of 2,3,7,8-TCDD in municipal and industrial discharges as providedunder 40 CFR Part 136.1. Method 625 may be used to screen samples for2,3,7,8-TCDD. When the screening test is positive, the final qualitative confirmationand quantification must be made using Method 613.11.3The method detection limit (MDL, defined in Section 14.1) for 2,3,7,8-TCDD is listedin Table 1. The MDL for a specific wastewater may be different from that listed,depending upon the nature of interferences in the sample matrix.1.4Because of the extreme toxicity of this compound, the analyst must prevent exposureto himself, of to others, by materials knows or believed to contain 2,3,7,8-TCDD.Section 4 of this method contains guidelines and protocols that serve as minimumsafe-handling standards in a limited-access laboratory.1.5Any modification of this method, beyond those expressly permitted, shall beconsidered as a major modification subject to application and approval of alternatetest procedures under 40 CFR Parts 136.4 and 136.5.1.6This method is restricted to use by or under the supervision of analysts experiencedin the use of a gas chromatograph/mass spectrometer and in the interpretation ofmass spectra. Each analyst must demonstrate the ability to generate acceptable results with this method using the procedure described in Section 8.2.2.Summary of Method2.1 A measured volume of sample, approximately 1 L, is spiked with an internal standardof labeled 2,3,7,8-TCDD and extracted with methylene chloride using a separatoryfunnel. The methylene chloride extract is exchanged to hexane during concentration to a volume of 1.0 mL or less. The extract is then analyzed by capillary columnGC/MS to separate and measure 2,3,7,8-TCDD.2,32.2The method provides selected column chromatographic cleanup proceudres to aid inthe elimination of interferences that may be encountered.3.Interferences3.1Method interferences may be caused by contaminants in solvents, reagents, glassware,and other sample processing hardware that lead to discrete artifacts and/or elevated backgrounds at the masses (m/z) monitored. All of these materials must be routinely demonstrated to be free from interferences under the conditions of the analysis byrunning laboratory reagent blanks as described in Section 8.1.3.43.1.1Glassware must be scrupulously cleaned. Clean all glassware as soon aspossible after use by rinsing with the last solvent used in it. Solvent rinsingshould be followed by detergent washing with hot water, and rinses with tapwater and distilled water. The glassware should then be drained dry, andheated in a muffle furnace at 400°C for 15-30 minutes. Some thermally stablematerials, such as PCBs, may not be eliminated by the treatment. Solventrinses with acetone and pesticide quality hexane may be substituted for themuffle furnace heating. Thorough rinsing with such solvents usuallyeliminates PCB interference. Volumetric ware should not be heated in a mufflefurnace. After drying and cooling, glassware should be sealed and stored in aclean environment to prevent any accumulation of dust or other contaminants.Store inverted or capped with aluminum foil.3.1.2The use of high purity reagents and solvents helps to mininmize interferenceproblems. Purification of solvents by distillation in all-glass systems may berequired.3.2Matrix interferences may be caused by contaminants that are coextracted from thesample. The extent of matrix interferences will vary considerably from source tosource, depending upon the nature and diversity of the industrial complex ormunicipality being sampled. 2,3,7,8-TCDD is often associated with other interferingchlorinated compounds which are at concentrations several magnitudes higher thanthat of 2,3,7,8-TCDD. The cleanup producers in Section 11 can be used to overcomemany of these interferences, but unique samples may require additional cleanup1,5-7approaches to eliminate false positives and achieve the MDL listed in Table 1.3.3The primary column, SP-2330 or equivalent, resolves 2,3,7,8-TCDD from the other 21TCDD insomers. Positive results using any other gas chromatographic column must be confirmed using the primary column.4.Safety4.1The toxicity or carcinogenicity of each reagent used in this method has not beenprecisely defined; however, each chemical compound should be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be reduced to the lowest possible level by whatever means available. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safehandling of the chemicals specified in this method. A reference file of material datahandling sheets should also be made available to all personnel involved in thechemical analysis. Additional references to laboratory safety are available and have8-10been identified for the information of the analyst. Benzene and 2,3,7,8-TCDD have been identified as suspected human or mammalian carcinogens.4.2Each laboratory must develop a strict safety program for handling 2,3,7,8-TCDD. Thefollowing laboratory practices are recommended:4.2.1Contamination of the laboratory will be minimized by conducting allmanipulations in a hood.4.2.2The effluents of sample splitters for the gas chromatograph and roughingpumps on the GC/MS should pass through either a column of activatedcharcoal or be bubbled through a trap containing oil or high-boiling alcohols.4.2.3Liquid waste should be dissolved in methanol or ethanol and irradiated withultraviolet light with a wavelength greater than 290 nm for several days. (UseF 40 BL lamps or equivalent). Analyze liquid wastes and dispose of thesolutions when 2,3,7,8-TCDD can no longer be detected.4.3Dow Chemical U.S.A. has issued the following precautimns (revised November 1978)for safe handling of 2,3,7,8-TCDD in the laboratory:4.3.1The following statements on safe handling are as complete as possible on thebasis of available toxicological information. The precautions for safe handlingand use are necessarily general in nature since detailed, specificrecommendations can be made only for the particular exposure andcircumstances of each individual use. Inquiries about specific operations oruses may be addressed to the Dow Chemical Company. Assistance inevaluating the health hazards of particular plant conditions may be obtainedfrom certain consulting laboratories and from State Departments of Health orof Labor, many of which have an industrial health service. 2,3,7,8-TCDD isextremely toxic to laboratory animals. However, it has been handled for yearswithout injury in analytical and biological laboratories. Techniques used inhandling radioactive and infectious materials are applicable to 2,3,7,8,-TCDD.4.3.1.1Protective equipment—Throw-away plastic gloves, apron or lab coat,safety glasses, and a lab hood adequate for radioactive work.4.3.1.2Training—Workers must be trained in the proper method of removingcontaminated gloves and clothing without contacting the exteriorsurfaces.4.3.1.3Personal hygiene—Thorough washing of hands and forearms after eachmanipulation and before breaks (coffee, lunch, and shift).4.3.1.4Confinement—Isolated work area, posted with signs, segregatedglassware and tools, plastic-backed absorbent paper on benchtops.4.3.1.5Waste—Good technique includes minimizing contaminated waste.Plastic bag liners should be used in waste cans. Janitors must betrained in the safe handling of waste.4.3.1.6Disposal of wastes—2,3,7,8-TCDD decomposes above 800°C. Low-levelwaste such as absorbent paper, tissues, animal remains, and plasticgloves may be burned in a good incinerator. Gross quantities(milligrams) should be packaged securely and disposed throughcommercial or governmental channels which are capable of handlinghigh-level radioactive wastes or extremely toxic wastes. Liquids shouldbe allowed to evaporate in a good hood and in a disposable container.Residues may then be handled as above.4.3.1.7Decontamination—For personal decontamination, use any mild soapwith plenty of scrubbing action. For decontamination of glassware,tools, and surfaces, Chlorothene NU Solvent (Trademark of the DowChemical Company) is the least toxic solvent shown to be effective.Satisfactory cleaning may be accomplished by rinsing with Chlorothene,then washing with any detergent and water. Dishwater may bedisposed to the sewer. It is prudent to minimize solvent wastesbecause they may require special disposal through commercial sourceswhich are expensive.4.3.1.8Laundry—Clothing known to be contaminated should be disposed withthe precautions described under Section 4.3.1.6. Lab coats or otherclothing worn in 2,3,7,8-TCDD work areas may be laundered.Clothing should be collected in plastic bags. Persons who convey thebags and launder the clothing should be advised of the hazard andtrained in proper handling. The clothing may be put into a washerwithout contact if the launderer knows the problem. The washershould be run through a cycle before being used again for otherclothing.4.3.1.9Wipe tests—A useful method of determining cleanliness of worksurfaces and tools is to wipe the surface with a piece of filter paper.Extraction and analysis by gas chromatography can achieve a limit ofsensitivity of 0.1 µg per wipe. Less than 1 µg of 2,3,7,8-TCDD persample indicates acceptable cleanliness; anything higher warrantsfurther cleaning. More than 10 µg on a wipe sample constitutes anacute hazard and requires prompt cleaning before further use of theequipment or work space. A high (10 µg) 2,3,7,8-TCDD level indicatesthat unacceptable work practices have been employed in the past.4.3.1.10Inhalation—Any procedure that may produce airborne contaminationmust be done with good ventilation. Gross losses to a ventilationsystem must not be allowed. Handling of the dilute solutions normallyused in analytical and animal work presents no inhalation hazardsexcept in the case of an accident.4.3.1.11Accidents—Remove contaminated clothing immediately, takingprecautions not to contaminate skin or other articles. Wash exposedskin vigorously and repeatedly until medical attention is obtained.5.Apparatus and Materials5.1Sampling equipment, for discrete or composite sampling.5.1.1Grab sample bottle—1 L or 1 qt, amber glass, fitted with a screw cap lined withTeflon. Foil may be substituted for Teflon if the sample is not corrosive. Ifamber bottles are not available, protect samples from light. The bottle and capliner must be washed, rinsed with acetone or methylene chloride, and driedbefore use to minimize contamination.5.1.2Automatic sampler (optional)—The sampler must incorporate glass samplecontainers for the collection of a minimum of 250 mL of sample. Samplecontainers must be kept refrigerated at 4°C and protected from light duringcompositing. If the sampler uses a peristaltic pump, a minimum length ofcompressible silicone rubber tubing may be used. Before use, however, thecompressible tubing should be thoroughly rinsed with methanol, followed byrepeated rinsings with distilled water to minimize the potential forcontamination of the sample. An integrating flow meter is required to collectflow proportional composites.5.1.3Clearly label all samples as “POISON” and ship according to U.S. Departmentof Transportation regulations.5.2Glassware (All specifications are suggested. Catalog numbers are included forillustration only)5.2.1Separatory funnels—2 L and 125 mL, with Teflon stopcock.5.2.2Concentrator tube, Kuderna-Danish—10 mL, graduated (Kontes K-570050-1025or equivalent). Calibration must be checked at the volumes employed in thetest. Ground glass stopper is used to prevent evaporation of extracts.5.2.3Evaporative flask, Kuderna-Danish—500 mL (Kontes K-570001-0500 orequivalent). Attach to concentrator tube with springs.5.2.4Snyder column, Kuderna-Danish—Three-ball macro (Kontes K-503000-0121 orequivalent).5.2.5Snyder column, Kuderna-Danish—Two-ball micro (Kontes K-569001-0219 orequivalent).5.2.6Vials—10-15 mL, amber glass, with Teflon-lined screw cap.5.2.7Chromatographic column—300 mm long x 10 mm ID, with Teflon stopcock andcoarse frit filter disc at bottom.5.2.8Chromatographic column—400 mm long x 11 mm ID, with Teflon stopcock andcoarse frit filter disc at bottom.5.3Boiling chips—Approximately 10/40 mesh. Heat to 400°C for 30 minutes or Soxhletextract with methylene chloride.5.4Water bath—Heated, with concentric ring cover, capable of temperature control(±2°C). The bath should be used in a hood.5.5GC/MS system5.5.1Gas chromatograph—An analytical system complete with a temperatureprogrammable gas chromatograph and all required accessories includingsyringes, analytical columns, and gases. The injection port must be designedfor capillary columns. Either split, splitless, or on-column injection techniquesmay be employed, as long as the requirements of Section 7.1.1 are achieved.5.5.2Column—60 m long x 0.25 mm ID glass or fused silica, coated with SP-2330 (orequivalent) with a film thickness of 0.2 µm. Any equivalent column mustresolve 2, 3, 7, 8-TCDD from the other 21 TCDD isomers.165.5.3Mass spectrometer—Either a low resolution mass spectrometer (LRMS) or ahigh resolution mass spectrometer (HRMS) may be used. The massspectrometer must be equipped with a 70 V (nominal) ion source and becapable of aquiring m/z abundance data in real time selected ion monitoring(SIM) for groups of four or more masses.5.5.4GC/MS interface—Any GC to MS interface can be used that achieves therequirements of Section 7.1.1. GC to MS interfaces constructed of all glass orglass-lined materials are recommended. Glass surfaces can be deactivated bysilanizing with dichlorodimethylsilane. To achieve maximum sensitivity, theexit end of the capillary column should be placed in the ion source. A shortpiece of fused silica capillary can be used as the interface to overcome problemsassociated with straightening the exit end of glass capillary columns.5.5.5The SIM data acquired during the chromatographic program is defined as theSelected Ion Current Profile (SICP). The SICP can be acquired under computercontrol or as a real time analog output. If computer control is used, there mustbe software available to plot the SICP and report peak height or area data forany m/z in the SICP between specified time or scan number limits.5.6Balance—Analytical, capable of accurately weighing 0.0001 g.6.Reagents6.1Reagent water—Reagent water is defined as a water in which an interferent is notobserved at the MDL of 2, 3, 7, 8-TCDD.6.2Sodium hydroxide solution (10 N)—Dissolve 40 g of NaOH (ACS) in reagent waterand dilute to 100 mL. Wash the solution with methylene chloride and hexane before use.6.3Sodium thiosulfate—(ACS) Granular.6.4Sulfuric acid—Concentrated (ACS, sp. gr. 1.84).6.5Acetone, methylene chloride, hexane, benzene, ortho-xylene, tetradecane—Pesticidequality or equivalent.6.6Sodium sulfate—(ACS) Granular, anhydrous. Purify by heating at 400°C for fourhours in a shallow tray.6.7Alumina—Neutral, 80/200 mesh (Fisher Scientific Co., No. A-540 or equivalent).Before use, activate for 24 hours at 130°C in a foil-covered glass container.6.8Silica gel—High purity grade, 100/120 mesh (Fisher Scientific Co., No. S-679 orequivalent).6.9Stock standard solutions (1.00 µg/µL)—Stock standard solutimns can be preparedfrom pure standard materials or purchased as certified solutions. Acetone should be used as the solvent for spiking solutions; ortho-xylene is recommended for calibration standards for split injectors; and tetradecane is recommended for splitless or on-colum injectors. Analyze stock internal standards to verify the absence of native2,3,7,8-TCDD.6.9.1Prepare stock standard solutions of 2,3,7,8-TCDD (mol wt 320) and either 37C142,3,7,8-TCDD (mol wt 328) or 13KC112K 2,3,7,8-TCDD (mol wt 332) in anisolated area by accurately weighing about 0.0100 g of pure material. Dissolvethe material in pesticide quality solvent and dilute to volume in a 10 mLvolumetric flask. When compound purity is assayed to be 96% or greater, theweight can be used without correction to calculate the concentration of thestock standard. Commercially prepared stock standards can be used at anyconcentration if they are certified by the manufacturer or by an independentsource.6.9.2Transfer the stock standard solutions into Teflon-sealed screw-cap bottles. Storein an isolated refrigerator protected from light. Stock standard solutions shouldbe checked frequently for signs of degradation or evaporation, especially justprior to preparing calibration standards or spiking solutions from them.6.9.3Stock standard solutions must be replaced after six months, or sooner ifcomparison with check standards indicates a problem.6.10Internal standard spiking solution (25 ng/mL)—Using stock standard solution,prepare a spiking solution in acetone of either13KCl12K or 37KCl4K 2,3,7,8-TCDD at a concentration of 25 ng/mL. (See Section 10.2)6.11Quality control check sample concentrate—See Section 8.2.1.7.Calibration 7.1Establish gas chromatograhic operating conditions equivalent to those given in Table 1and SIM conditions for the mass spectrometer as described in Section 12.2. TheGC/MS system must be calibrated using the internal standard technique.7.1.1Using stock standards, prepare calibration standards that will allowmeasurement of relative response factors of at least three concentration ratios of2,3,7,8-TCDD to internal standard. Each calibration standard must be preparedto contain the internal standard at a concentration of 25 ng/mL. If anyinterferences are contributed by the internal standard at m/z 320 and 322, itsconcentration may be reduced in the calibration standards and in the internalstandard spiking solution (Section 6.10). One of the calibration standardsshould contain 2,3,7,8-TCDD at a concentration near, but above, the MDL andthe other 2,3,7,8-TCDD concentrations should correspond to the expected rangeof concentrations found in real samples or should define the working range ofthe GC/MS system.7.1.2Using injections of 2-5 µL, analyze each calibration standardaccording toSection 12 and tabulate peak height or area response against the concentrationof 2,3,7,8-TCDD and internal standard. Calculate response factors (RF) for2,3,7,8-TCDD using Equation 1.Equation 1where:A = SIM response for 2,3,7,8-TCDD m/z 320.s A = SIM response for the internal standard, m/z 332 for C is 1213 2,3,7,8-TCDD m/z 328 for Cl 2,3,7,8-TCDD.374C = Concentration of the internal standard (µg/L).is C = Concentration of 2,3,7,8-TCDD (µg/L).s If the RF value over the working range is a constant (<10% relative standarddeviation, RSD), the RF can be assumed to be invariant and the average RF canbe used for calculations. Alternatively, the results can be used to plot acalibration curve of response ratios, A /A , vs. concentration ratios C /C .s is s is *7.1.3The working calibration curve or RF must be verified on each working day bythe measurement of one or more 2,3,7,8-TCDD calibration standards. If theresponse for 2,3,7,8-TCDD varies from the predicted response by more than±15%, the test must be repeated using a fresh calibration standard.Alternatively, a new calibration curve must be prepared.7.2Before using any cleanup procedure, the analyst must process a series of calibrationstandards through the procedure to validate elution patterns and the absence ofinterferences from the reagents.8.Quality Control8.1Each laboratory that uses this method is required to operate a formal quality controlprogram. The minimum requirements of this program consist of an initialdemonstration of laboratory capability and an ongoing analysis of spiked samples to evaluate and document data quality. The laboratory must maintain records todocument the quality of data that is generated. Ongoing data quality checks arecompared with established performance criteria to determine if the results of analyses meet the performance characteristics of the method. When results of sample spikesindicate atypical method performance, a quality control check standard must beanalyzed to confirm that the measurements were performed in an in-control mode of operation.8.1.1The analyst must make an initial, one-time, demonstration of the ability togenerate acceptable accuracy and precision with this method. This ability isestablished as described in Section 8.2.8.1.2In recognition of advances that are occurring in chromatography, the analyst ispermitted certain options (detailed in Sections 10.5, 11.1, and 12.1) to improvethe separations or lower the cost of measurements. Each time such amodification is made to the method, the analyst is required to repeat theprocedure in Section 8.28.1.3Before processing any samples, the analyst must analyze a reagent water blankto demonstrate that interferences from the analytical system and glassware areunder control. Each time a set of samples is extracted or reagents are changed,a reagent water blank must be processed as a safeguard against laboratorycontamination.8.1.4The laboratory must, on an ongoing basis, spike and analyze a minimum of10% of all samples with native 2,3,7,8-TCDD to monitor and evaluate laboratorydata quality. This procedure is described in Section 8.3.8.1.5The laboratory must, on an ongoing basis, demonstrate through the analyses ofquality control check standards that the operation of the measurement system isin control. This procedure is described in Section 8.4. The frequency of thecheck standard analyses is equivalent to 10% of all samples analyzed but maybe reduced if spike recoveries from samples (Section 8.3) meet all specifiedquality control criteria.8.1.6The laboratory must maintain performance records to document the quality ofdata that is generated. This procedure is described in Section 8.5.8.2To establish the ability to generate acceptable accuracy and precision, the analyst mustperform the following operations.8.2.1 A quality control (QC) check sample concentrate is required containing2,3,7,8-TCDD at a concentration of 0.100 µg/mL in acetone. The QC checksample concentrate must be obtained from the U.S. Environmental ProtectionAgency, Environmental Monitoring and Support Laboratory in Cincinnati, Ohio,if available. If not available from that source, the QC check sample concentratemust be obtained from another external source. If not available from eithersource above, the QC check sample concentrate must be prepared by thelaboratory using stock standards prepared independently from those used forcalibration.8.2.2Using a pipet, prepare QC check samples at a concentration of 0.100 µg/L(100 ng/L) by adding 1.00 mL of QC check sample concentrate to each of four1 L aliquots of reagent water.8.2.3Analyze the well-mixed QC check samples according to the method beginningin Section 10.8.2.4recovery (s) in µg/L, for 2,3,7,8-TCDD using the four results.8.2.5) with the corresponding acceptance criteria for precision andcriteria, the system performance is acceptable and analysis of actual samplesaccuracy, the system performance is unacceptable for 2,3,7,8-TCDD. Locate andcorrect the source of the problem and repeat the test beginning withSection 8.2.2.8.3The laboratory must, on an ongoing basis, spike at least 10% of the samples from eachsample site being monitored to assess accuracy. For laboratories analyzing one to ten samples per month, at least one spiked sample per month is required.8.3.1The concentration of the spike in the sample should be determined as follows:8.3.1.1If, as in compliance monitoring, the concentration of 2,3,7,8-TCDD inthe sample is being checked against a regulatory concentration limit,the spike should be at that limit or one to five times higher than thebackground concentration determined in Section 8.3.2, whicheverconcentration would be larger.8.3.1.2If the concentration of 2,3,7,8-TCDD in the sample is not being checkedagainst a limit specific to that parameter, the spike should be at0.100 µg/L or one to five times higher than the backgroundconcentration determined in Section 8.3.2, whichever concentrationwould be larger.8.3.1.3If it is impractical to determine background levels before spiking (e.g.,maximum holding times will be exceeded), the spike concentrationshould be (1) the regulatory concentration limit, if any; or, if none(2) the larger of either five times higher than the expected backgroundconcentration or 0.100 µg/L.8.3.2Analyze one sample aliquot to determine the background concentration (B) of2,3,7,8-TCDD. If necessary, prepare a new QC check sample concentrate(Section 8.2.1) appropriate for the background concentration in the sample.Spike a second sample aliquot with 1.0 mL of the QC check sample concentrateand analyze it to determine the concentration after spiking (A) of 2,3,7,8-TCDD.Calculate percent recovery (P) as 100 (A-B)%T, where T is the known true valueof the spike.8.3.3Compare the percent recovery (P) for 2,3,7,8-TCDD with the corresponding QCacceptance criteria found in Table 2. These acceptance criteria were calculatedto include an allowance for error in measurement of both the background andspike concentrations, assuming a spike to background ratio of 5:1. This errorwill be accounted for to the extent that the analyst's spike to background ratio11approaches 5:1. If spiking was performed at a concentration lower than0.100 µg/L, the analyst must use either the QC acceptance criteria in Table 2, oroptional QC acceptance criteria calculated for the specific spike concentration.To calculate optional acceptance criteria for the recovery of 2,3,7,8-TCDD:(1) Calculate accuracy (X′) using the equation in Table 3, substituting the spike′) using the equation inTable 3, substituting X′concentration as (100 X′/T) ±2.44(100 S′/T)%.118.3.4If the recovery of 2,3,7,8-TCDD falls outside the designated range for recovery,a check standard must be analyzed as described in Section 8.4.8.4If the recovery of 2,3,7,8-TCDD fails the acceptance criteria for recovery in Section 8.3,a QC check standard must be prepared and analyzed.NOTE:The frequency for the required analysis of a QC check standard willdepend upon the complexity of the sample matrix and the performance ofthe laboratory.8.4.1Prepare the QC check standard by adding 1.0 mL of QC check sampleconcentrate (Section 8.2.1 or 8.3.2) to 1 L of reagent water.8.4.2Analyze the QC check standard to determine the concentration measured (A) of2,3,7,8-TCDD. Calculate the percent recovery (P K) as 100 (A/T)%, where T issthe true value of the standard concentration.8.4.3Compare the percent recovery (P K) with the corresponding QC acceptancescriteria found in Table 2. If the recovery of 2,3,7,8-TCDD falls outside thedesignated range, the laboratory performance is judged to be out of control, andthe problem must be immediately identified and corrected. The analytical。
美国环保局 EPA 试验 方法9071bn-Hexane Extractable Material (HEM) for Sludge, Sediment, and Solid S

Internet9071B - 1Revision 2April 1998METHOD 9071Bn-HEXANE EXTRACTABLE MATERIAL (HEM) FOR SLUDGE, SEDIMENT, AND SOLID SAMPLES 1.0SCOPE AND APPLICATION1.1Method 9071 may be used to quantify low concentrations of oil and grease in soil,sediments, sludges, and other solid materials amenable to chemical drying and solvent extraction with n-hexane. “Oil and grease” is a conventional pollutant under 40 CFR 401.16 and generally refers to substances, including biological lipids and mineral hydrocarbons, that have similar physical characteristics and common solubility in an organic extracting solvent. As such, oil and grease is an operationally defined parameter, and the results will depend entirely on the extracting solvent and method of extraction. Method 9071 employs n-hexane as the extraction solvent with Soxhlet extraction and the results of this method are appropriately termed “n-hexane extractable material (HEM).” Section 1.2 lists the type of materials that may be extracted by this method. In the context of this method, “HEM” is used throughout this method and for operational purposes, may be considered synonymous with “oil and grease” within the limitations discussed below.1.2Specifically, Method 9071 is suitable for extracting relatively non-volatile hydrocarbons,vegetable oils, animal fats, waxes, soaps, greases, biological lipids, and related materials. 1.3Method 9071 is not recommended for measuring materials that volatilize at temperatures below 85E C. Petroleum fuels from gasoline through #2 fuel oil may be partially lost during the solvent removal process.1.4 Some crude oils and heavy fuel oils may contain materials that are not soluble in n-hexane, and recovery of these materials may be low.2.0SUMMARY OF METHOD2.1 A representative portion of wet (as received) waste is acidified with concentrated HCl and chemically dried with magnesium sulfate or sodium sulfate. Magnesium sulfate monohydrate is used to dry acidified sludges as it will combine with 75% of its own weight in water in forming MgSO C 7H O. Anhydrous sodium sulfate is used to dry soil and sediment samples. 4 2 2.2After drying, the HEM is extracted with n-hexane using a Soxhlet apparatus. The n-hexane extract is then distilled from the extract and the HEM is desiccated and weighed. 2.3 When necessary, a separate sample portion is evaluated for percent solids, and the dry weight fraction may be used to calculate the dry-weight HEM concentration of the soil, sediment,or waste.3.0DEFINITIONS3.1n-Hexane extractable material (HEM, oil and grease): Material that is extracted from a sample using n-hexane and determined by this method. This material includes relatively non-volatile hydrocarbons, vegetable oils, animal fats, waxes, soaps, greases, and related matter.3.2Refer to Chapter One for additional definitions.4.0INTERFERENCES4.1 This method is entirely empirical, and duplicate results having a high degree of precision can be obtained only by strict adherence to all details. The rate of cycling and time of extraction in the Soxhlet apparatus must be consistent and length of time required for drying and cooling extracted materials must be the same in order to generate consistent results. It is important that the procedures be performed as directed due to the varying solubilities of the different greases and heavy mineral oils.4.2Solvents, reagents, glassware, and other sample-processing hardware may yield artifacts that could affect the results. All solvents and reagents used in the analysis should be demonstrated to be free from interferences by processing a method blank with each analytical batch. Specific selection of reagents, solvent washes, or purification of solvents may be required. Use of plastic measuring devices, and/or plastic tubing attachments must be avoided.4.3Glassware should be cleaned by washing with hot tap water with detergent, rinsing with tap water and reagent water, and rinsing with solvent. Glassware may also be baked at 200-250E C for 1 hour. Boiling flasks that are used to contain the extracted residues may be dried in an oven at 105-115E C and stored in a desiccator until used. Depending on the project DQOs, strict adherence to the washing and handling procedures cited above may not be necessary as long as the laboratory can demonstrate that alternative cleaning procedures yield acceptable method performance and meet method blank acceptance criteria.4.4 A gradual increase in weight may result due to the absorption of oxygen; a gradual loss of weight may result due to volatilization. Extracted residues should be maintained in a desiccator during cooling and prior to weighing. Extracted residues should be weighed as soon as possible after cooling.4.5The presence of non-oily extractable substance such as sulfur compounds, organic dyes, and chlorophyll, may result in a positive bias. For the purpose of this method, all materials extracted and retained during this procedure are defined as HEM.5.0SAFETY5.1The toxicity or carcinogenicity of each reagent used in this method has not been precisely determined; however, each chemical should be treated as a potential health hazard. Exposure to these chemicals should be reduced to the lowest possible level. It is suggested that the laboratory perform personal hygiene monitoring of each analyst that uses this method. This monitoring should be performed using Occupational Safety and Health Administration (OSHA) or National Institute of Occupational Safety and Health (NIOSH) approved personal hygiene monitoring methods. Results of this monitoring should be made available to the analyst.5.2n-Hexane has been shown to have increased neurotoxic effects over other hexanes and some other solvents. OSHA has proposed a time-weighted average (TWA) of 50 parts-per-million (ppm); NIOSH concurs that an 8-hour TWA/permissible exposure limit (PEL) of 50 ppm is appropriate for n-hexane; and the American Conference of Governmental Industrial Hygienists (ACGIH) has published a threshold limit value (TLV) of 50 ppm for n-hexane. Inhalation of n-hexane should be minimized by performing all operations with n-hexane in a explosion-proof hood or well-ventilated area.Internet9071B - 2Revision 2April 19985.3n-Hexane has a flash point of -23E C (-9E F), has explosive limits in air in the range of 1 to 7 percent, and poses a serious fire risk when heated or exposed to flame. n-Hexane can react vigorously with oxidizing materials. The laboratory should include procedures in its operations that address the safe handling of n-hexane.5.4Unknown samples may contain high concentrations of volatile toxic compounds. Sample containers should be opened in a hood and handled with gloves to prevent exposure.5.5This method does not address all safety issues associated with its use. The laboratory is responsible for maintaining a safe work environment and a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets (MSDSs) should be available to all personnel involved in these analyses.6.0EQUIPMENT AND SUPPLIES6.1Soxhlet extraction apparatus.6.2Heating mantle - explosion-proof, with temperature control.6.3Boiling flask - 125-mL or appropriate size.6.4Analytical balance - capable of weighing 0.1 mg.6.5Vacuum pump, or other vacuum source.6.6Paper extraction thimble for Soxhlet apparatus.6.7Glass wool or small glass beads to fill thimble.6.8Grease-free, non-absorbent cotton - To remove possible interferences, each batch of cotton should be washed with n-hexane. Solvent washing may not be necessary if the laboratory can demonstrate that the unwashed cotton does not affect the performance of the method or that the concentration of HEM in the sample is so high that low contaminant concentration is insignificant.6.9Beakers - 100- 150-mL.6.10pH paper.6.11Porcelain mortar and pestle.6.12Extraction flask - 150-mL or appropriate size.6.13Waterbath or steam bath-explosion-proof - capable of maintaining a temperature of at least 85E C.6.14Distilling apparatus - For removing n-hexane from extract.6.14.1Distilling head-Claisen (VWR Scientific No 26339-005, or equivalent), includesClaisen-type connecting tube and condenser.Internet9071B - 3Revision 2April 1998Internet9071B - 4Revision 2April 19986.14.2Distillation adapter (used to attach distilling head and to the waste collectionflask for recovery of solvent).6.14.3Distillate collection flask (attached to the distilling adaptor for collection of thedistilled solvent).6.14.4Ice bath or recirculating chiller (to aid in the condensation and collection ofthe distilled solvent).6.15Desiccator - Cabinet or jar type, capable of holding boiling flasks during cooling and storage.6.16Tongs - for handling the boiling flasks.6.17Glass fiber filter paper - Whatman No. 40 or equivalent.6.18Boiling chips - Silicon carbide or fluoropolymer.7.0REAGENTS7.1Reagent grade chemicals shall be used in all tests. Unless otherwise indicated, it is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Other grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lessening the accuracy of the determination.7.2Reagent water. All references to water in this method refer to reagent water, as defined in Chapter One.7.3Concentrated hydrochloric acid (HCl).7.4 Magnesium sulfate monohydrate. Prepare MgSO C H O by spreading a thin layer in 4 2a dish and drying in an oven at 150E C overnight. Store in a tightly sealed glass container until used.7.5Sodium sulfate, granular, anhydrous (Na SO ). Purify by heating at 400E C for 4 hours 24in a shallow tray, or by precleaning the sodium sulfate with methylene chloride. If the sodium sulfate is precleaned with methylene chloride, a method blank must be analyzed, demonstrating that there is no interference from the sodium sulfate. Store in a tightly sealed glass container until used.7.6n-Hexane. Purity of 85%, 99.0% minimum saturated C isomers, residue less than 16mg/L. Boiling point, 69E C.7.7Hexadecane(CH (CH )CH )/stearic acid (CH (CH )COOH). 1:1 spiking solution.32143 3216Prepare in acetone at a concentration of 2 mg/mL each.Weigh 200 ± 2 mg of stearic acid and 200 ± 2 mg hexadecane into a 100 mL volumetric flask and fill to the mark with acetone. The total concentration of this stock is 4000 mg/L (ppm) HEM. This standard may be used for spiking samples and preparing laboratory control samples. Store in a glass container with a fluoropolymer-lined cap at room temperature. Shield from light.Note:The spiking solution may require warming for complete dissolution of stearic acid.8.0SAMPLE COLLECTION, PRESERVATION, AND STORAGE8.1 A minimum of 100 grams of sample should be collected using a metal spatula, spoon, or equivalent device. Samples should be collected into a pre-cleaned wide-mouth glass container fitted with a TFE-lined screw cap.8.2 When practical (i.e., when the sample matrix allows the complete mixing of sample and acid such as with a pourable sludge or sediment), the sample should be preserved to a pH < 2 by adding 1 mL of concentrated HCl per 100 gram of sample and cooled to 4 ± 2 E C. If acidification is not practical (as with a dry soil), the addition of the HCl is not required and the sample should be cooled to 4 ± 2 E C. The laboratory must be notified so that the sample can be acidified prior to analysis.8.3 A holding time has not been established for HEM in solids, but it is recommended that the sample be analyzed as soon as possible.9.0QUALITY CONTROL9.1Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and the analysis of spiked samples as a continuing check on performance. The laboratory is required to maintain performance records to define the quality of data that is generated.9.2 Employ a minimum of one method blank per analytical batch or twenty samples, whichever is more frequent, to verify that all reagents, solvents, and equipment are contamination free. Prepare the method blank from 5 g of inert matrix such as pre-cleaned sand or similar material, and carry it through the analytical process.9.3 Run one matrix duplicate and matrix spike sample every twenty samples or analytical batch, whichever is more frequent. Matrix duplicates and spikes are brought through the whole sample preparation and analytical process.9.4The performance of the method should be evaluated by the use of a Laboratory Control Sample (LCS). The LCS is prepared by spiking an inert matrix (as pre-cleaned sand or similar material) with an appropriate volume of spiking solution (Sec. 7.7) and carrying it through the analytical process.10.0CALIBRATION AND STANDARDIZATION10.1Calibrate the analytical balance at 2 mg and 1000 mg using class “S” weights.10.2Calibration shall be within ± 10% (i.e., ± 0.2 mg) at 2 mg and ± 0.5 % (i.e., ± 5 mg) at 1000 mg. If values are not within these limits, recalibrate the balance.Internet9071B - 5Revision 2April 1998dry weight fraction 'g of dry sample g of sampleInternet 9071B - 6Revision 2April 199811.0PROCEDURE11.1Determination of Sample Dry Weight Fraction11.1.1When it is necessary to report the HEM on a dry weight basis, determine thedry weight fraction using a separate aliquot of sample, as discussed below. The aliquot used for this determination cannot be used to evaluate HEM.11.1.2Weigh 5-10 gram (± 0.01 gram) of the sample into pre-weighed crucible.Determine the weight of the wet sample by subtracting the weight of the crucible.11.1.3Place the crucible with the wet sample in an oven overnight at 105E C.Remove crucible from oven and place in a desiccator to cool. Weigh. Determine dry weight of sample by subtracting the weight of the crucible. Determine the dry weight fraction of the sample as follows:NOTE:The drying oven should be contained in a hood or vented. Significant laboratory contamination may result from a heavily contaminated hazardous waste sample.11.2Sample Preparation 11.2.1Sludge/Waste Samples 11.2.1.1Weigh out 20 ± 0.5 grams of wet sample into a 150-mL beaker.11.2.1.2If the sample has not been acidified, acidify to a pH # 2 withapproximately 0.3 mL concentrated HCl.11.2.1.3Add 25 grams Mg SO C H O (Sec. 7.4) and stir to a smooth24 2paste.11.2.1.4 Spread paste on sides of beaker to facilitate evaporation. Letstand about 15-30 min or until material is solidified.11.2.1.5 Remove solids and grind to fine powder in a mortar.11.2.1.6Add the powder to the paper extraction thimble.11.2.1.7 Wipe beaker and mortar with pieces of filter paper moistened withn-hexane and add to thimble.11.2.1.8Fill thimble with glass wool (or glass beads).11.2.2Sediment/Soil Samples11.2.2.1Decant and discard any water layer on a sediment sample. Mixsample thoroughly, especially composited samples. Discard any foreign objects suchas sticks, leaves, and rocks.11.2.2.2Blend 10 grams of the sample with 10 grams of anhydrous sodiumsulfate (Sec. 7.5) as described in Section 11.2.1. Transfer homogenized paste to anextraction thimble and cover with glass wool or glass beads. The extraction thimblemust drain freely for the duration of the extraction period.11.3Extraction11.3.1 Set-up the Soxhlet apparatus containing the extraction thimble and sampleand attach a 125-mL boiling flask containing 90 mL of n-hexane. Add boiling chips. Adjust the heating control on the heating mantle so that a cycling rate of 20 cycles/h is obtained. Extract for a period of 4 hrs.11.3.2Tare a clean 250-mL or appropriate sized boiling flask as follows:11.3.2.1Dry the flask in an oven at 105-115E C for a minimum of 2 h.11.3.2.2Remove from the oven and immediately transfer to a desiccatorto cool at room temperature.11.3.2.3When cool, remove from the desiccator with tongs and weighimmediately on a calibrated balance.11.3.3At the end of the 4 h extraction period, filter the extract through grease-freecotton, into the pre-weighed boiling flask (Sec. 11.3.2). Use gloves to avoid adding fingerprints to the flask.11.3.4Rinse flask and cotton with n-hexane and add to the 250-mL boiling flask.NOTE:If the extract is clear and no suspended particles are present, the filtration step may be omitted.11.3.5Connect the boiling flask to the distilling head apparatus and distill the solventby immersing the lower half of the flask in a water bath or a steam bath. A heating mantle may also be used. Adjust the temperature of the heating device to complete the distillation in less than 30 minutes. Collect the solvent for reuse or appropriate disposal.11.3.6When the distillation is complete, remove the distilling head. Immediatelyremove the flask from the heat source and wipe the outside to remove excess moisture and fingerprints. To remove solvent vapor, sweep out the flask for 15 sec with air by inserting a glass tube that is connected to a vacuum source.11.3.7Cool the boiling flask in a desiccator for 30 min and weigh. Determine thegain in weight of the boiling flask by subtracting the weight of the boiling flask (Sec. 11.3.2) from the final boiling flask weight.Internet9071B - 7Revision 2April 1998HEM (mg/kg wet weight)'gain in weight of flask(mg)X 1000weight of wet solid(g)Internet9071B - 8Revision 2April 199812.0DATA ANALYSIS AND CALCULATIONSCalculate the concentration of HEM in the sample as follows:NOTE:If it is necessary to report the results on a dry weight basis, divide the result obtained above by the dry weight fraction calculated in Sec. 11.1.3. Report the results as mg/kg HEM dry weight. If it is necessary to report the results as a percentage of the wet or dry weight, divide the wet-weight concentration or dry weight concentration by 10,000 and report the result as % HEM wet or dry weight.13.0METHOD PERFORMANCEIn a preliminary study designed to find a suitable replacement for Freon-113, three EPA contract laboratories evaluated a total of 28 solid samples derived from various industrial and commercial processes for oil and grease. This study evaluated a total of six solvents, including n-hexane, to determine which of the alternative solvents produced results most closely with that of Freon-113. In this study, each waste was Soxhlet-extracted in triplicate using Freon-113 and each of the alternative solvents. Based on the overall results, n-hexane was judged to be the best alternative solvent. The data provided in Table 1 compare the results for Freon-113 and n-hexane for each waste. For a complete discussion of this study, refer to reference 1 in Section 16.0.14.0POLLUTION PREVENTION14.1Pollution prevention encompasses any technique that reduces or eliminates the quantity and/or toxicity of waste at the point of generation. Numerous opportunities for pollution prevention exist in laboratory operation. The EPA has established a preferred hierarchy of environmental management techniques that places pollution prevention as the management option of first choice.Whenever feasible, laboratory personnel should use pollution prevention techniques to address their waste generation. When wastes cannot be feasibly reduced at the source, the Agency recommends recycling as the next best option.14.2For information about pollution prevention that may be applicable to laboratories and research institutions consult Less is Better: Laboratory Chemical management for Waste Reduction available from the American Chemical Society’s Department of Government Relations and Science Policy, 1155 16th St., N.W. Washington, D.C. 20036, (202) 872-4477.15.0WASTE MANAGEMENTThe Environmental Protection Agency requires that laboratory waste management practices be conducted consistent with all applicable Federal, state and local rules and regulations. The Agency urges laboratories to protect the air, water, and land by minimizing and controlling all releases from hoods and bench operations, complying with the letter and spirit of any sewerdischarge permits and regulations, and by complying with all solid and hazardous waste regulations, particularly the hazardous waste identification rules and land disposal restrictions. For further information on waste management, consult The Waste Management Manual for Laboratory Personnel available from the American Chemical Society at the address listed in Sec. 14.2.16.0REFERENCES1.Preliminary Report of EPA Efforts to Replace Freon for the Determination of Oil and Grease,United States Environmental Protection Agency, Office of Water, EPA-821-93-009. June 1993.2.Method 1664, Revision A: n-Hexane Extractable Material (HEM; Oil and Grease) and Silica GelTreated N-Hexane Extractable Material (SGT-HEM) by Extraction and Gravimetry.17.0TABLES, DIAGRAMS, FLOWCHARTS, AND VALIDATION DATAThe pages to follow contain Table 1, and a flow diagram of the method procedure.Internet9071B - 9Revision 2April 1998TABLE 1SOXHLET EXTRACTION OF SOLIDS USING FREON-113 AND N-HEXANEAll concentrations in mg/kgFacility/Waste Stream Solvent:Rep Rep Rep Mean Standard Process Freon No. 1 No. 2No. 3Concen-DeviationHexane trationPaper Mill Dewatered Freon110005300790080002762 Sludge Hexane660024001100066004203POTW Sewage Freon980008100081000870009940 Sludge Hexane11000086000800009100013281Leather Dewatered Freon11000120001200012000732 Tannery Sludge Hexane210001500019000180003201 POTW Digested Freon13000097000660009800033028 Sludge Hexane5400076000480005900014516Petroleum API Separator Freon32000035000025000031000053257 Refinery Sludge Hexane24000032000024000027000043822 Industrial DAF Freon31000031000024000029000041717 Laundry Sludge Hexane29000036000018000028000090819Fish Oil Oily Freon8900001000000770000890000131249 Plant Sludge Hexane44000053000046000048000046318 Coke Plant Waste Freon8300800018000110005505 Activated Hexane140001900015000160002732SludgeWood Solid Freon1500001400001400001400003512 Preserving Waste Hexane1400001300001300001300006557 PlantDrilling Fluid Used drilling Freon1300160013001400157 Supplier mud Hexane1300120016001400201 Contam.Kerosene Freon2000140019001700352 Soils Contaminated Hexane2500320026002800410 SoilPoultry Plant Waste Freon3800011000400003000016263 Activated Hexane590011000460002100021795SludgeRolling Mill Dewatered Freon110001400017000140002884 Scale Hexane14000140001600015000983 Mayonnaise Oily Sludge Freon88000085000078000084000050521 Plant Hexane590000780000520000630000132020Seafood Waste Sludge Freon6400053000580007526 Plant Hexane340003100027000310003867 Internet9071B - 10Revision 2April 1998TABLE 1(CONTINUED)Facility/Waste Stream Solvent:Rep Rep Rep Mean Standard Process Freon No. 1 No. 2No. 3Concen-DeviationHexane trationSeafood Oily Sludge Freon40000041000043000041000016371 Plant Hexane4000003900003900004000007095Poultry DAF Sludge Freon67000060000057000061000049549 Plant Hexane5300005300005300005300002449Railroad Oily Sludge Freon87000092000087000089000027906 Yard Hexane8500008400008300008400006884 Can Filter Cake Freon62000620006000061000976 Manufact Hexane690006400066000660002615 PlantSoup Plant DAF Sludge Freon60000059000061000060000010066Hexane58000052000060000057000040361Oily Water Oily Sludge Freon760007500070000740003215 Treatment Hexane7700060000790007200010713 PlantCan Oily Sludge Freon940008800094000920003291 Manufact Hexane800009000083000850004992 PlantCan Filter Cake Freon2900002900003000002900006217 Manufact Hexane2900002900002900002900002029 PlantDrum Oily Sludge Freon120000011000001200000120000057735 Handling Hexane9900001000000980000100000027319 FacilityPolymer Dewatered Freon13000120008200110002524 Plant Sludge Hexane84006900910081001122 Restaurant Vegetable Oil Freon76000061000078000072000092060 Waste Hexane1100000980000980000100000080064Leather Waste Sludge Freon18000022000019000019000022140 Tannery Hexane24000027000021000024000031177 Source: Reference 1Internet9071B - 11Revision 2April 1998METHOD 9071Bn-HEXANE EXTRACTABLE MATERIAL (HEM) FOR SLUDGE, SEDIMENT, AND SOLID SAMPLESInternet9071B - 12Revision 2April 1998METHOD 9071Bn-HEXANE EXTRACTABLE MATERIAL (HEM) FOR SLUDGE, SEDIMENT, AND SOLID SAMPLES(Continued)Internet9071B - 13Revision 2April 1998。
美国饮用水水质标准(EPA)

《美国饮用水水质标准》(EPA)
[标题]:《美国饮用水水质标准》
[颁布者]:美国
[编号]:
[颁布日期]:
[实施日期]:
[有效性]:有效
国家一级饮用水规程(NPDWRs或一级标准),是法定强制性的标准,它适用于公用给水系统。
一级标准限制了那些有害公众健康的及已知的或在公用给水系统中出现的有害污染物浓度,从
注:
①、污染物最高浓度目标MCLG-对人体健康无影响或预期无不良影响的水中污染物浓度。
它规定了确当的安全限量,MCLGs是非强制性公共健康目标。
②、污染物最高浓度-它是供给用户的水中污染物最高允许浓度,MCLGs它是强制性标准,MCLG 是安全限量,确保略微超过MCL限量时对公众健康不产生显着风险。
③、TT处理技术-公共给水系统必须遵循的强制性步骤或技术水平以确保对污染物的控制。
④、除非有特别注释,一般单位为mg/L。
⑤、
度:
病毒
HPC每毫升不超过500细菌数。
⑨、每月总大肠杆菌阳性水样不超过5%,于每月例行检测总大肠杆菌的样品少于40只的给水系统,总大肠菌阳性水样不得超过1个。
含有总大肠菌水样,要分析粪型大肠杆菌,粪型大肠杆菌不容许存在。
⑩、粪型及艾氏大肠杆菌的存在表明水体受到人类和动物排泄物的污染,这些排泄物中的微生物可引起腹泻,痉挛,恶心,头痛或其它症状。
岳宇明译岳舜琳校。
EPA方法索引

EPA方法索引和相关标准品EPA 是美国国家环境保护局(U.S Environmental Protection Agency) 的英文缩写。
它的主要任务是保护人类健康和自然环境。
EPA 制定了一系列标准分析方法用于环境监测领域。
主要包括:EPA T01~T14 系列标准分析方法——空气中有毒有机物分析方法EPA IP1~IP10 系列标准分析方法——室内空气污染物的分析测定方法EPA 200 系列标准分析方法———金属的分析方法EPA 500 系列标准分析方法——饮用水中有机物的分析方法EPA 600 系列标准分析方法——城市和工业废水中有机化合物的分析方法SW -846 系列标准分析方法——固体废弃物试验分析评价手册1300 系列是毒性试验方法3000 系列是金属元素的提取方法3500 系列是半(非) 挥发性有机物的提取方法3600 系列是净化、分离方法5000 系列是挥发性有机物的提取方法6000 系列是测定金属的新方法7000 系列是原子吸收法测定金属元素8000 系列是有机物分析方法9000 系列是常规项目分析方法其中,500系列,600系列和8000系列是环境种有机物分析最常用的方法。
EPA 600系列方法是美国为贯彻“净水法”(CW A) 、“全国水体污染物排放消除制度”(NPDES) 和“许可证制度”,严格控制点源排放,保护地表水,使其免受城市和工业废水中有机物的污染而制定的。
EPA 500 系列方法是为执行“安全饮用水法”(SDW A) 和“国家一级饮用水法案”(National Primary Drinking Water Regulations) ,确保饮用水及饮用水源的质量而制订的。
EPA 500 系列是针对比较洁净的水样(饮用水、地下水、地表水) 开发的,有些方法仅用试剂水和饮用水验证过SW-846 系列集中贯彻了“资源保护回收法”和“陆地处置限制法规”的精神,包含了固体废弃物采样和分析试验的全部方法, 是在EPA200 ~EPA 600 系列的基础上发展起来的。
美国EPA 关于空气自动监测系统性能指标的规定和测试方法
美国EPA关于大气自动监测系统性能指标的规定和测试方法引言环境空气污染的自动监测方法有多种,一般采用湿法和干法两种。
湿法是基于化学量理论的库仑法和电导法等测量原理,需使用大量试剂,存在试剂调整和废液处理等问题,操作比较繁琐,故障率较高,维护工作量较大;干法是基于物理光谱测量理论,使样品始终保持在气体状态,没有试剂的损耗,维护工作量较小。
比如SO2测量采用紫外荧光法,NOx测量采用化学发光法,O3测量采用紫外光度法,CO测量采用气体过滤相关分析法等,目前我国绝大部分空气自动监测采用的是该方法。
干法测量以欧美为主。
美国开展空气自动监测已有30年的历史,在空气自动监测方面积累了丰富的经验,并制定了详细的规范。
其中物理光谱法作为美国EPA的推荐方法,得到了广泛的应用。
湿法测量以日本为主,但自1996年起日本在法定的测量方法中增加了干式测量法。
利用物质的光谱特性进行污染物的分析已成为自动监测仪器发展的必然趋势。
我国在环境空气质量监测和质量保证方面的规定都参考了美国国家环保署(EPA)的规定。
目前,大气自动监测和空气质量日报工作在我国大部分省市已广泛开展,自动监测仪器监测数据的准确可靠是日报工作中的基础。
为使监测人员了解美国EPA关于空气自动监测的相关规定,特将其有关SO2、NO2、O3、CO自动监测仪器的性能指标规定和测试方法作简要说明,以供参考。
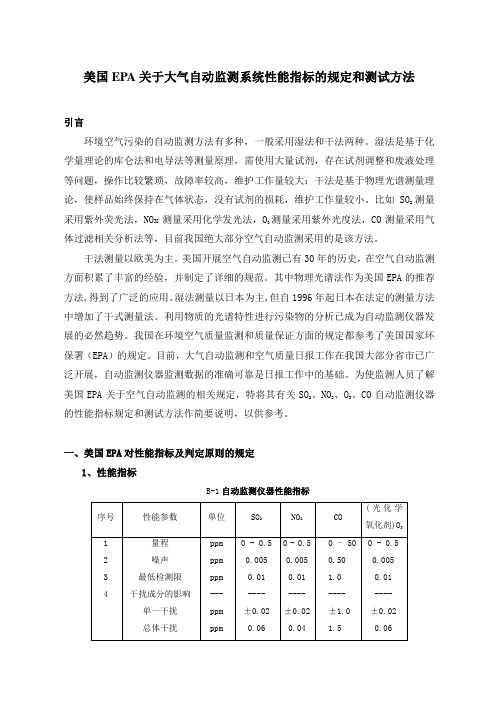
一、美国EPA对性能指标及判定原则的规定1、性能指标B-1自动监测仪器性能指标M/0.02447,M是该气体的摩尔质量。
2、判定原则对于每个性能指标(量程除外),测试程序从开始起要重复7次,得到7组测试结果。
每组结果要和表B-1中的规定指标相比较,高于或超出规定指标的值是一个超标值。
每个参数的7个结果说明如下:(1)0次超标:被测的参数合格;(2)3次或更多次超标:该参数不合格;(3)1次或2次超标:再重复测试该参数 8次,得到共15个测试结果。
将此15个测试结果说明如下:a:1次或2次超标:通过测试;b:3次以上:该参数不合格。
美国EPA最新参考方法标准

特别规定的样品采集过滤器。
手动参考方法: 配备 RAAS-10 PM10 进气口或
RFPS-0699-131 40 联邦法规(CFR)第 50 部分,
附录 L, 图 L-2 到 L-19 中特定的
联邦公告:卷 64, 有通气孔的进口,作为 PM10
第 33481 页 , 参考方法配置,流量为 16.67 升
图 L-2 参考方法
第 33481 页 , 配置,流量为 16.67 升/分钟,24
BGI 公司 BGI 公司 DKK-TOA 公司 Ecotech 公司
PQ100 型空气采样器
PQ200 型空气采样器
FPM-222/222C,FPM223 /223C 及 DUB-222(S)型 PM10 监测器 3000 型 PM10 大容量空 气采样器
或
12/01/87 及卷 53, GMW-IP-10-8000 中的任一型号
第 1062 页 , 大容量采样器,这些采样器含有
01/15/88
以下部件:带有丙烯腈-丁二烯-
苯乙烯塑胶过滤器托架和电机/
鼓风机外壳或不锈钢过滤器托
架和酚醛塑料电机/鼓风机外壳
的阳极氧化处理铝制大容量外
壳;0.6 大功率电机/鼓风机; 压
06/23/99
小时连续采样周期操作。符合
RAAS105-300 操作说明书,遵
循 40 CFR 第 50 部分,附录 J
或附录 M 中有关要求和特别规
定的样品采集过滤器。
手动参考方法: 配备 BGI16.7 进气口装置或附
RFPS-0699-132 录 L,40 联邦法规(CFR)50,
图 L-2 到 L-19 中特定的有通气
7.0 说明书,适当的还带有特制
EPA方法索引

EPA方法索引EPA(Environmental Protection Agency,环境保护局)是美国联邦政府机构,负责制定环境保护政策和监督执行,旨在保护人类健康和自然环境。
EPA通过开发和更新一系列的方法和准则来评估和监测环境中的各种污染物。
以下是EPA方法的索引,其中包含了一些常用的方法。
1.水质分析方法:-EPA方法6010:使用电感耦合等离子体质谱仪对水样中的重金属进行测定。
-EPA方法160.2:测定饮用水中总溶解性氟化物的浓度。
-EPA方法200.7:使用火焰原子吸收光谱法测定水样中的金属。
-EPA方法365.2:测定地下水中40种有机化合物的浓度。
2.大气质量监测方法:-EPA方法305:测定大气中颗粒物(PM10)的质量浓度。
-EPA方法1664:对水和底泥中的油脂进行提取和测定。
-EPA方法321.8:通过气浓度梯度法测定大气中的苯系化合物。
-EPA方法327:使用红外光谱法测定大气中的多环芳烃。
3.土壤和底泥分析方法:-EPA方法3540:对土壤和底泥中的有机物进行提取。
-EPA方法8000:使用气相色谱质谱法分析土壤和底泥中的挥发性有机化合物。
-EPA方法3051:测定土壤样品中重金属的浓度。
-EPA方法8240:使用气相色谱质谱法分析土壤和底泥中的半挥发性有机化合物。
4.垃圾和固体废物分析方法:-EPA方法8015:使用气相色谱质谱法分析固体废物中的多环芳烃。
-EPA方法8082:使用气相色谱质谱法分析土壤、底泥和固体废物中的戴奥辛和类似化合物。
-EPA方法8260:使用气相色谱质谱法分析固体废物中的挥发性有机化合物。
-EPA方法8280:使用气相色谱质谱法分析固体废物中的多氯联苯。
5.生物监测方法:-EPA方法1600:测定饮用水和海水中的大肠杆菌和肠球菌数量。
-EPA方法1613:使用液相色谱质谱法测定鱼类组织中的多氯联苯和多溴联苯醚。
-EPA方法2050:测定水和生物体中蓝绿藻的数量和类群组成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
METHOD 9012BTOTAL AND AMENABLE CYANIDE (AUTOMATED COLORIMETRIC,WITH OFF-LINE DISTILLATION)1.0 SCOPE AND APPLICATION1.1 This method is used to determine the concentration of inorganic cyanide (CAS Registry Number 57-12-5) in wastes or leachate. This method detects inorganic cyanides that are present as either soluble salts or complexes. It is used to determine values for both total cyanide and cyanide amenable to chlorination. The "reactive" cyanide content of a waste is not determined by this method. Refer to 40 CFR 261.23 for information on the characteristic of reactivity.2.0 SUMMARY OF METHOD2.1 The cyanide, as hydrocyanic acid (HCN), is released from samples containing cyanide by means of a reflux-distillation operation under acidic conditions and absorbed in a scrubber containing sodium hydroxide solution. The cyanide ion in the absorbing solution is then determined by automated UV colorimetry.2.2 In the automated colorimetric measurement, the cyanide is converted to cyanogen chloride (CNCl) by reaction with Chloramine-T at a pH less than 8 without hydrolyzing to the cyanate. After the reaction is complete, color is formed on the addition of pyridine-barbituric acid reagent. The concentration of NaOH must be the same in the standards, the scrubber solutions, and any dilution of the original scrubber solution to obtain colors of comparable intensity.3.0 INTERFERENCES3.1Interferences are eliminated or reduced by using the distillation procedure. Chlorine and sulfide are interferences in this method.3.2Oxidizing agents such as chlorine decompose most cyanides. Chlorine interferences can be removed by adding an excess of sodium arsenite to the waste prior to preservation and storage of the sample to reduce the chlorine to chloride which does not interfere.3.3Sulfide interference can be removed by adding an excess of bismuth nitrate to the waste (to precipitate the sulfide) before distillation. Samples that contain hydrogen sulfide, metal sulfides, or other compounds that may produce hydrogen sulfide during the distillation should be treated by the addition of bismuth nitrate.3.4High results may be obtained for samples that contain nitrate and/or nitrite. During the distillation, nitrate and nitrite will form nitrous acid, which will react with some organic compounds to form oximes. These compounds once formed will decompose under test conditions to generate HCN. The possibility of interference of nitrate and nitrite is eliminated by pretreatment with sulfamic acid just before distillation. Nitrate and nitrite are interferences when present at levels higher than 10 mg/L and in conjunction with certain organic compounds.3.5Thiocyanate is reported to be an interference when present at very high levels. Levels of 10 mg/L were not found to interfere in Method 9010.3.6Fatty acids, detergents, surfactants, and other compounds may cause foaming during the distillation when they are present in large concentrations and will make the endpoint of the titration difficult to detect. They may be extracted at pH 6-7.4.0 APPARATUS AND MATERIALS4.1Reflux distillation apparatus such as shown in Figure 1 or Figure 2. The boiling flask should be of one liter size with inlet tube and provision for condenser. The gas scrubber may be a 270-mL Fisher-Milligan scrubber (Fisher, Part No. 07-513 or equivalent). The reflux apparatus may be a Wheaton 377160 distillation unit or equivalent.4.2 Automated continuous-flow analytical instrument with:4.2.1 Sampler.4.2.2 Manifold.pump.4.2.3 Proportioning4.2.4 Heating bath with distillation coil.4.2.5 Distillationhead.4.2.6 Colorimeter equipped with a 15-mm flowcell and 570 nm filter.4.2.7 Recorder.4.3Hot plate stirrer/heating mantle.4.4pH meter.4.5Amber light.4.6Vacuum source.4.7Refrigerator.4.8 5 mL microburette.4.97 Class A volumetric flasks -- 100 and 250 mL.4.10Erlenmeyer flask -- 500 mL.5.0 REAGENTS5.1Reagent grade chemicals shall be used in all tests. Unless otherwise indicated, it is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Othergrades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lessening the accuracy of the determination.5.2Reagent water. All references to water in this method refer to reagent water, as defined in Chapter One.5.3Reagents for sample collection, preservation, and handling 5.3.1Sodium arsenite (0.1N), NaAsO 2. Dissolve 3.2 g of NaAsO 2 in 250 mL water.5.3.2Ascorbic acid, C 6H 8O 6.5.3.3Sodium hydroxide solution (50%), NaOH. Commercially available.5.3.4Acetic acid (1.6M) CH 3COOH. Dilute one part of concentrated acetic acidwith 9 parts of water.5.3.52,2,4-Trimethylpentane, C 8H 18.5.3.6Hexane, C 6H 14.5.3.7Chloroform, CHCl 3.5.4Reagents for cyanides amenable to chlorination5.4.1Calcium hypochlorite solution (0.35M), Ca(OCl)2. Combine 5 g of calciumhypochlorite and 100 mL of water. Shake before using.5.4.2Sodium hydroxide solution (1.25N), NaOH. Dissolve 50 g of NaOH in 1liter of water.5.4.3Sodium arsenite (O.1N). See Sec. 5.3.1.5.4.4Potassium iodide starch paper.5.5Reagents for distillation5.5.1Sodium hydroxide (1.25N). See Sec. 5.4.2.5.5.2Bismuth nitrate (0.062M), Bi(NO)3 C 5H 2O. Dissolve 30 g of Bi(NO)3 C5H 2O in 100 mL of water. While stirring, add 250 mL of glacial acetic acid, CH 3COOH. Stir until dissolved and dilute to 1 liter with water.5.5.3Sulfamic acid (0.4N), H 2NSO 3H. Dissolve 40 g H 2NSO 3H in 1 liter ofwater. 5.5.4Sulfuric acid (18N), H 2SO 4. Slowly and carefully add 500 mL ofconcentrated H 2SO 4 to 500 mL of water.5.5.5Magnesium chloride solution (2.5M), MgCl 2 C 6H 2O. Dissolve 510 g ofMgCl 2 C 6H 2O in 1 liter of water.5.5.6Lead acetate paper.5.6Reagents for automated colorimetric determination5.6.1Pyridine-barbituric acid reagent -- Place 15 g of barbituric acid in a 250-mL volumetric flask, add just enough reagent water to wash the sides of the flask, and wet the barbituric acid. Add 75 mL of pyridine and mix. Add 15 mL of concentrated HCl, mix, and cool to room temperature. Dilute to 250 mL with reagent water and mix. This reagent is stable for approximately six months if stored in a cool, dark place.5.6.2Chloramine-T solution -- Dissolve 2.0 g of white, water solublechloramine-T in 500 mL of reagent water and refrigerate until ready to use.5.6.3Sodium hydroxide, 1 N -- Dissolve 40 g of NaOH in reagent water, anddilute to 1 liter.5.6.4All working standards should contain 2 mL of 1 N NaOH (Sec. 5.6.3) per100 mL.5.6.5Dilution water and receptacle wash water (NaOH, 0.25 N) -- Dissolve 10.0g of NaOH in 500 mL of reagent water. Dilute to 1 liter.6.0 SAMPLE COLLECTION, PRESERVATION, AND HANDLING6.1Samples should be collected in plastic or glass containers. All containers must be thoroughly cleaned and rinsed.6.2Oxidizing agents such as chlorine decompose most cyanides. To determine whether oxidizing agents are present, test a drop of the sample with potassium iodide-starch test paper. A blue color indicates the need for treatment. Add 0.1N sodium arsenite solution a few mL at a time until a drop of sample produces no color on the indicator paper. Add an additional 5 mL of sodium arsenite solution for each liter of sample. Ascorbic acid can be used as an alternative although it is not as effective as arsenite. Add a few crystals of ascorbic acid at a time until a drop of sample produces no color on the indicator paper. Then add an additional 0.6 g of ascorbic acid for each liter of sample volume.6.3Aqueous samples must be preserved by adding 50% sodium hydroxide until the pH is greater than or equal to 12 at the time of collection.6.4Samples should be chilled to 4 E C.6.5When properly preserved, cyanide samples can be stored for up to 14 days prior to sample preparation steps.6.6Solid and oily wastes may be extracted prior to analysis by Method 9013 (Cyanide Extraction Procedure for Solids and Oils). It uses a dilute NaOH solution (pH = 12) as the extractant. This yields extractable cyanide.6.7If fatty acids, detergents, and surfactants are a problem, they may be extracted using the following procedure. Acidify the sample with acetic acid (1.6M) to pH 6.0 to7.0. CAUTION:This procedure can produce lethal HCN gas.Extract with isooctane, hexane, or chloroform (preference in order named) with solvent volume equal to 20% of the sample volume. One extraction is usually adequate to reduce thecompounds below the interference level. Avoid multiple extractions or a long contact time at low pH in order to keep the loss of HCN at a minimum. When the extraction is completed,immediately raise the pH of the sample to above 12 with 50% NaOH solution.7.0 PROCEDURE7.1Pretreatment for cyanides amenable to chlorination7.1.1This test must be performed under amber light. K 3[Fe-(CN)6] maydecompose under UV light and hence will test positive for cyanide amenable tochlorination if exposed to fluorescent lighting or sunlight. Two identical sample aliquots are required to determine cyanides amenable to chlorination.7.1.2To one 500 mL sample or to a sample diluted to 500 mL, add calciumhypochlorite solution dropwise while agitating and maintaining the pH between 11 and 12with 1.25N sodium hydroxide until an excess of chlorine is present as indicated by KI-starch paper turning blue. The sample will be subjected to alkaline chlorination by this step. CAUTION: The initial reaction product of alkaline chlorination is the very toxic gascyanogen chloride; therefore, it is necessary that this reaction beperformed in a hood.7.1.3Test for excess chlorine with KI-starch paper and maintain this excess forone hour with continuous agitation. A distinct blue color on the test paper indicates a sufficient chlorine level. If necessary, add additional calcium hypochlorite solution.7.1.4After one hour, add 1 mL portions of 0.1N sodium arsenite until KI-starchpaper shows no residual chlorine. Add 5 mL of excess sodium arsenite to ensure the presence of excess reducing agent.7.1.5Test for total cyanide as described below in both the chlorinated and theunchlorinated samples. The difference of total cyanide in the chlorinated andunchlorinated samples is the cyanide amenable to chlorination.7.1.6If samples are known or suspected to contain sulfide, add 50 mL of0.062M bismuth nitrate solution through the air inlet tube. Mix for three minutes. Use lead acetate paper to check the sample for the presence of sulfide. A positive test is indicated by a black color on the paper.7.2Distillation procedure7.2.1Place 500 mL of sample, or sample diluted to 500 mL in the one literboiling flask. Pipet 50 mL of 1.25N sodium hydroxide into the gas scrubber. If theapparatus in Figure 1 is used, add water until the spiral is covered. Connect the boiling flask, condenser, gas scrubber and vacuum trap.7.2.2Start a slow stream of air entering the boiling flask by adjusting thevacuum source. Adjust the vacuum so that approximately two bubbles of air per second enter the boiling flask through the air inlet tube.7.2.3If samples are known or suspected to contain nitrate or nitrite, or if bismuth nitrate was added to the sample, add 50 mL of 0.4N sulfamic acid solution through the air inlet tube. Mix for three minutes.NOTE: Excessive use of sulfamic acid could create method bias.7.2.4Slowly add 50 mL of 18N sulfuric acid through the air inlet tube. Rinse the tube with water and allow the airflow to mix the flask contents for three minutes. Add 20 mL of 2.5M magnesium chloride through the air inlet and wash the inlet tube with a stream of water.7.2.5Heat the solution to boiling. Reflux for one hour. Turn off heat and continue the airflow for at least 15 min. After cooling the boiling flask, and closing the vacuum source, disconnect the gas scrubber.7.2.6Transfer the solution from the scrubber into a 250-mL volumetric flask. Rinse the scrubber into the volumetric flask. Dilute to volume with water.7.3 Automated colorimetric determination7.3.1 Set up the manifold in a hood or a well-ventilated area as shown in Figure 3.7.3.2 Allow colorimeter and recorder to warm up for 30 min. Run a baseline with all reagents, feeding reagent water through the sample line.7.3.3 Place appropriate standards in the sampler in order of increasing concentration. Complete loading of the sampler tray with unknown samples.7.3.4 When the baseline becomes steady, begin the analysis.7.4Standard curve for samples without sulfide7.4.1Prepare a series of standards by pipetting suitable volumes of working standard potassium cyanide solution into 250-mL volumetric flasks. To each flask, add 50 mL of 1.25N sodium hydroxide and dilute to 250 mL with water. Prepare using the following table. The sodium hydroxide concentration will be 0.25N.mL of Working Standard Solution(1 mL = 10 µg CN-)Concentration (µg CN-/L)0.01.02.0 5.0 10.0 15.0 20.0Blank 40 80 200 400 600 8007.4.2After the standard solutions have been prepared according to the table above, pipet 50 mL of each standard solution into a 100-mL volumetric flask and proceedto Secs. 7.3.2 and 7.3.3 to obtain absorbance values for the standard curve. The finalconcentrations for the standard curve will be one half of the amounts in the above table (final concentrations ranging from 20 to 400 µg/L).7.4.3It is recommended that at least two standards (a high and a low) bedistilled and compared to similar values on the curve to ensure that the distillationtechnique is reliable. If distilled standards do not agree within ± 10% of the undistilledstandards, the analyst should find the cause of the apparent error before proceeding.7.4.4Prepare a standard curve ranging from 20 to 400 µg/L by plottingabsorbance of standard versus the cyanide concentration7.5Standard curve for samples with sulfide7.5.1It is imperative that all standards be distilled in the same manner as thesamples using the method of standard additions (for example, bismuth nitrate must also be added to the standards). Standards distilled by this method will give a linear curve, at low concentrations, but as the concentration increases, the recovery decreases. It isrecommended that at least five standards be distilled.7.5.2Prepare a series of standards similar in concentration to those mentionedin Sec. 7.4.1 and analyze as in Sec. 7.3. Prepare a standard curve by plotting absorbance of standard versus the cyanide concentration.7.6 Calculation -- Prepare a standard curve by plotting peak heights of standards against their concentration values. Compute concentrations of samples by comparing sample peak heights with the standard curve.8.0 QUALITY CONTROL8.1Refer to Chapter One for specific quality control procedures.8.2Verify the calibration curve with an independent calibration check standard. If the standards are not within 15% of the expected value, a new recalibration curve is required. Verify the calibration curve with every sample batch by analyzing a mid-range standard.8.3Run one matrix spike sample for every 10 samples to check the efficiency of sample distillation. A matrix spike should be prepared by adding cyanide from the working standard or intermediate standard to 500 mL of sample to ensure a concentration of approximately 40 µg/L. Both the matrix duplicate and matrix spike duplicate are brought through the entire sample preparation and analytical process.8.4 The method of standard additions shall be used for the analysis of all samples that suffer from matrix interferences such as samples which contain sulfides.9.0 METHOD PERFORMANCE9.1 Precision and accuracy data are not available at this time.10.0REFERENCES1. Annual Book of ASTM Standards, Part 31, "Water," Standard D2036-75, Method B, p. 505 (1976).2. Goulden, P.D., B.K. Afghan, and P. Brooksbank, Determination of Nanogram Quantities of Simple and Complex Cyanides in Water, Anal. Chem., 44(11), pp. 1845-49 (1972).3. Standard Methods for the Examination of Water and Wastewater, 14th ed., pp. 376 and 370, Method 413F and D (1975).4. Technicon AutoAnalyzer II Methodology, Industrial Method No. 315-74 WCUV Digestion and Distillation, Technicon Industrial Systems, Tarrytown, New York, 10591 (1974).FIGURE 1APPARATUS FOR CYANIDE DISTILLATIONFIGURE 2CYANIDE DISTILLATION APPARATUSFIGURE 3 CYANIDE MANIFOLD AA11METHOD 9012BTOTAL AND AMENABLE CYANIDE (AUTOMATED COLORIMETRICWITH OFF-LINE DISTILLATION )METHOD 9012B (continued)。