美国环保局 EPA 试验 方法 3520c
二氯硫化碳检测标准

二氯硫化碳检测标准二氯硫化碳(CS2)是一种有机化合物,其检测通常涉及空气、水或土壤中的含量。
对于不同的环境介质,可能存在不同的检测标准和方法。
以下是一些常见的检测标准和方法:1.空气中的检测:•美国职业安全与健康管理局(OSHA):OSHA规定了空气中二氯硫化碳的允许接触限值(Permissible ExposureLimit,PEL),以确保工作场所空气中的CS2浓度在安全范围内。
PEL通常以时间加权平均(Time-WeightedAverage,TWA)和短期曝露限值(Short-Term ExposureLimit,STEL)表示。
•美国国家职业安全卫生研究所(NIOSH):NIOSH也提供了关于二氯硫化碳的安全指南和检测方法。
2.水中的检测:•美国环境保护署(EPA):EPA规定了水中二氯硫化碳的最大容许浓度,以确保饮用水和环境水体中的CS2浓度处于安全水平。
•国际标准化组织(ISO):ISO可能也提供了一些关于水质标准和检测方法的国际标准。
3.土壤中的检测:•美国环境保护署(EPA):EPA可能规定了土壤中二氯硫化碳的安全限值和检测方法,以确保土壤中的CS2浓度对人类和环境没有危害。
4.其他国家和地区的标准:•根据不同国家和地区的法规和标准,对于二氯硫化碳的检测和限值可能存在差异。
因此,在具体的应用中,应该参考相应国家或地区的法规和标准。
需要注意的是,CS2的检测方法通常使用气相色谱法(Gas Chromatography,GC)等分析技术。
实际的检测程序和标准可能根据不同的监管机构和研究机构而有所不同。
在进行检测之前,建议咨询专业的环境监测和分析服务提供商。
EN及EPA等系列标准 中文名称

BS EN1122:2001:湿法消解测定塑料中的镉BS EN 1122:2001:Plastics-determination of cadmium-Wet decomposition method方法3005A:FLAA,ICP方法分析酸式消解水中的总溶解金属METHOD 3005A:ACID DIGESTION OF WATERS FOR TOTAL RECVERABLE OR DISSOLVED METALS FOR ANALYSIS BY FLAA OR ICP SPECTROSCOPY方法3010A: FLAA,ICP方法分析酸式消解水样和蒸馏样中的总金属METHOD 3010A:ACID DIGESTION OF AQUEOUS SAMPLES AND EXTRACTS FOR TOTAL METALS FOR ANALYSIS BY FLAA OR ICP SPECTROSCOPY方法3015:微波酸式消解水样和蒸馏液METHOD 3015:MICROWAVE ASSISTED ACID DIGESTION OF AQUEOUS SAMPLES AND EXTRACTS 方法3020A:G FAA方法分析酸式消解水样和蒸馏样中的总金属METHOD 3010A:ACID DIGESTION OF AQUEOUS SAMPLES AND EXTRACTS FOR TOTAL METALS FOR ANALYSIS BY GFAA SPECTROSCOPY方法3031:AAS,ICP分析酸式消解石油中的金属元素METHOD 3031:ACID DIGESTION OF OILS FOR METALS ANALYSIS BY ATOMIC ABSORPTION OR ICP SPECTROMETRY方法3040A:石油,油脂,石蜡的消解程序METHOD 3040A:DISSOLUTION PROCEDURE FOR OILS, GREASES, OR WAXES方法3050B:沉淀物,淤泥,土壤的酸式消解METHOD 3050B:ACID DIGESTION OF SEDIMENTS, SLUDGES, AND SOILS方法3051:沉淀物,淤泥,土壤石油的微波酸式消解METHOD 3051:MICROWAVE ASSISTED ACID DIGESTION OF SEDIMENTS SLUDGES, SOILS, AND OILS方法3052:硅酸盐和有机质的微波酸式消解METHOD 3052:MICROWAVE ASSISTED ACID DIGESTION OF SILICEOUS AND ORGANICALLY BASED MATRICES方法3060:六价铬的碱式消解METHOD 3060A:ALKALINE DIGESTION FOR HEXAVALENT CHROMIUM方法3500B:有机萃取和样品制备METHOD 3500B:ORGANIC EXTRACTION AND SAMPLE PREPARATION方法3510C:分液漏斗的液—液萃取METHOD 3510C :SEPARATORY FUNNEL LIQUID-LIQUID EXTRACTION方法3520C:连续液-液萃取METHOD3520C:CONTINUOUS LIQUID-LIQUID EXTRACTION方法3535:固相萃取METHOD 3535:SOLID-PHASE EXTRACTION (SPE)方法3540C:索氏萃取SOXHLET EXTRACTION方法3541:AUTOMATED SOXHLET EXTRACTION方法3542:用方法0010收集半挥发性分析物的萃取物METHOD 3542:EXTRACTION OF SEMIVOLATILE ANALYTES COLLECTED USING METHOD 0010(MLDIFIED METHOD 5 SAMPLING TRAIN)方法3545:密闭流动萃取METHOD 3545:PRESSURIZED FLUID EXTRACTION (PFE)方法3546:微波萃取METHOD 3546:MICROWAVE EXTRACTION方法3550B:超声波萃取METHOD 3550B:ULTRASONIC EXTRACTION方法3560:超临界流动萃取石油中的中回收物METHOD 3560:SUPERCRITICAL FLUID EXTRACTION OF TOTAL RECOVERABLE PETROLEUM HYDROCARBONS方法3561:多环芳烃的超临界流动萃取METHOD 3561:SUPERCRITICAL FLUID EXTRACTION OF POLYNUCLEAR AROMATIC HYDROCARBONS方法3580:废物稀释METHOD 3580A:WASTE DILUTION方法3585:挥发性有机物的废物稀释METHOD 3585:WASTE DILUTION FOR VOLATILE ORGANICS方法3600C:清除METHOD 3600C:CLEANUP方法3610B:矾土的清除METHOD 3610B:ALUMINA CLEANUP方法3611B:柱状矾土的清除和石油废弃物的分离METHOD 3611B:ALUMINA COLUMN CLEANUP AND SEPARATION OF PETROLEUM WASTES方法3520B:硅酸镁的清除METHOD 3520B:FLORISIL CLEANUP方法3630C:硅胶的清除METHOD 3630C:SILICA GEL CLEANUP方法3640A:渗入硅胶的清除METHOD 3640A:GEL-PERMEATION CLEANUP方法3650B:酸碱的分离清除METHOD 3650B:ACID-BASE PARTITION CLEANUP方法3660B:硫磺的清除METHOD3660B:SULFUR CLEANUP方法3665A:硫酸,高锰酸的清除METHOD 3665A:SULFURIC ACID/PERMANGANATE CLEANUP方法3810:顶部空间METHOD 3810:HEADSPACE方法3820:16烷萃取和净化有机物的屏蔽METHOD 3820:HEXADECANE EXTRACTION AND SCREENING OF PURGEABLE ORGANICS方法7000A:原子吸收方法METHOD 7000A:ATOMIC ABSORPTION METHODS方法7130:镉(原子吸收,直接吸收)METHOD7130:CADMIUM (ATOMIC ABSORPTION, DIRECT ASPIRATION)方法7130A:镉(原子吸收,炉子技术)METHOD7131A:CADMIUM (ATOMIC ABSORPTION, FURNACE TECHNIQUE)方法7190:铬(原子吸收,直接吸收)METHOD7190:CHROMIUM (ATOMIC ABSORPTION, DIRECT ASPIRATION)方法7191:铬(原子吸收,炉子技术)METHOD7191:CHROMIUM (ATOMIC ABSORPTION, FURNACE TECHNIQUE)方法7196A:六价铬(比色)METHOD 7196A:CHROMIUM, HEXAVALENT (COLORIMETRIC)方法7420:铅(原子吸收,直接吸收)方法7420:LEAD (ATOMIC ABSORPTION, DIRECT ASPIRATION)方法7421:铅(原子吸收,炉子技术)METHOD 7421:LEAD (ATOMIC ABSORPTION, DIRECT ASPIRATION)方法7470A:废水中的汞(冷原子蒸汽技术)METHOD 7470A:MERCURY IN LIQUID WASTE (MANUAL COLD-VAPOR TECHNIQUE)方法7471A:固体和固体废弃物中的汞(冷原子蒸汽技术)METHOD 7471A:MERCURY IN SOLID OR SEMISOLID WASTE (MANUAL COLD-VAPOR TECHNIQUE) 方法7473 :热分解原子吸收光谱法测定固体和液体样品中的汞METHOD 7473:MERCURY IN SOLIDS AND SOLUTIONS BY THERMAL DECOMPODITION AMALGAMATION, AND ATOMIC ABSORPTION SPECTROPHOTOMETRY方法8000B:限定色谱分离METHOD 8000B:DETERMINATIVE CHROMATOGRAPHIC SEPARATIONS方法8081A:气相色谱分析有机氯沙虫剂METHOD 8081A:ORGANOCHLORINE PESTICIDES BY GAS CHROMATOGRAPHY方法8081B:气相色谱分析有机氯沙虫剂METHOD 8081B:ORGANOCHLORINE PESTICIDES BY GAS CHROMATOGRAPHY方法8082:气相色谱分析多氯联苯METHOD 8082:POLYCHLORINATED BIPHENYLS (PCBs) BY GAS CHROMATOGRAPHY方法8082A:气相色谱分析多氯联苯METHOD 8082A:POLYCHLORINA BIPHENYLS (PCBs) BY GAS CHROMATOGRAPHY方法8260B:气相色谱/质谱分析挥发性有机化合物METHOD 8260B:VOLATILE ORGANIC COMPOUNDS BY GAS CHROMATOGRAPHY/MASS SPECTROMETRY (GC/MS)。
epa standard method 533

epa standard method 533
EPA(美国环境保护局)Standard Method 533是关于气体监测的方法,具体名为《Method 533:Determination of Volatile Organic Compounds (VOCs) in Air by Adsorption on Activated Carbon and Gas Chromatography》。
这个方法主要用于测定空气中的挥发性有机化合物(VOCs)。
在这个方法中,样品通过吸附在活性炭上收集,然后使用气相色谱法进行分析。
方法涵盖了吸附剂的选择、采样设备、样品处理和分析等步骤。
活性炭吸附剂具有较高的吸附能力,可以有效地捕获空气中的VOCs。
气相色谱法用于分离和定量吸附剂上的VOCs,从而得出空气中VOCs的浓度。
EPA Standard Method 533为监测空气中的挥发性有机化合物提供了可靠的方法,有助于评估空气质量并制定相应的环境保护措施。
该方法在我国环保领域也有广泛应用,以保障空气质量和人民健康。
EPA-Method-3501[1]
![EPA-Method-3501[1]](https://img.taocdn.com/s3/m/3da4fd8ecc22bcd126ff0c4a.png)
DETERMINATION OF AMMONIA NITROGEN BY SEMI-AUTOMATEDCOLORIMETRYEdited by James W. O'DellInorganic Chemistry BranchChemistry Research DivisionRevision 2.0August 1993ENVIRONMENTAL MONITORING SYSTEMS LABORATORY OFFICE OF RESEARCH AND DEVELOPMENTU.S. ENVIRONMENTAL PROTECTION AGENCYCINCINNATI, OHIO 45268350.1-1DETERMINATION OF AMMONIA NITROGEN BY SEMI-AUTOMATEDCOLORIMETRY1.0SCOPE AND APPLICATION1.1This method covers the determination of ammonia in drinking, ground,surface, and saline waters, domestic and industrial wastes.1.2The applicable range is 0.01-2.0 mg/L NH as N. Higher concentrations can be3determined by sample dilution. Approximately 60 samples per hour can beanalyzed.1.3This method is described for macro glassware; however, micro distillationequipment may also be used.2.0SUMMARY OF METHOD2.1The sample is buffered at a pH of 9.5 with a borate buffer in order to decreasehydrolysis of cyanates and organic nitrogen compounds, and is distilled into asolution of boric acid. Alkaline phenol and hypochlorite react with ammoniato form indophenol blue that is proportional to the ammonia concentration.The blue color formed is intensified with sodium nitroprusside and measuredcolorimetrically.2.3Reduced volume versions of this method that use the same reagents and molarratios are acceptable provided they meet the quality control and performancerequirements stated in the method.2.4Limited performance-based method modifications may be acceptable providedthey are fully documented and meet or exceed requirements expressed inSection 9.0, Quality Control.3.0DEFINITIONS3.1Calibration Blank (CB) -- A volume of reagent water fortified with the samematrix as the calibration standards, but without the analytes, internalstandards, or surrogate analytes.3.2Calibration Standard (CAL) -- A solution prepared from the primary dilutionstandard solution or stock standard solutions and the internal standards andsurrogate analytes. The CAL solutions are used to calibrate the instrumentresponse with respect to analyte concentration.350.1-23.3Instrument Performance Check Solution (IPC) -- A solution of one or moremethod analytes, surrogates, internal standards, or other test substances usedto evaluate the performance of the instrument system with respect to a definedset of criteria.3.4Laboratory Fortified Blank (LFB) -- An aliquot of reagent water or other blankmatrices to which known quantities of the method analytes are added in thelaboratory. The LFB is analyzed exactly like a sample, and its purpose is todetermine whether the methodology is in control, and whether the laboratoryis capable of making accurate and precise measurements.3.5Laboratory Fortified Sample Matrix (LFM) -- An aliquot of an environmentalsample to which known quantities of the method analytes are added in thelaboratory. The LFM is analyzed exactly like a sample, and its purpose is todetermine whether the sample matrix contributes bias to the analytical results.The background concentrations of the analytes in the sample matrix must bedetermined in a separate aliquot and the measured values in the LFMcorrected for background concentrations.3.6Laboratory Reagent Blank (LRB) -- An aliquot of reagent water or other blankmatrices that are treated exactly as a sample including exposure to allglassware, equipment, solvents, reagents, internal standards, and surrogatesthat are used with other samples. The LRB is used to determine if methodanalytes or other interferences are present in the laboratory environment, thereagents, or the apparatus.3.7Linear Calibration Range (LCR) -- The concentration range over which theinstrument response is linear.3.8Material Safety Data Sheet (MSDS) -- Written information provided byvendors concerning a chemical's toxicity, health hazards, physical properties,fire, and reactivity data including storage, spill, and handling precautions.3.9Method Detection Limit (MDL) -- The minimum concentration of an analytethat can be identified, measured and reported with 99% confidence that theanalyte concentration is greater than zero.3.10Quality Control Sample (QCS) -- A solution of method analytes of knownconcentrations that is used to fortify an aliquot of LRB or sample matrix. TheQCS is obtained from a source external to the laboratory and different fromthe source of calibration standards. It is used to check laboratory performancewith externally prepared test materials.3.11Stock Standard Solution (SSS) -- A concentrated solution containing one ormore method analytes prepared in the laboratory using assayed referencematerials or purchased from a reputable commercial source.4.0INTERFERENCES350.1-34.1Cyanate, which may be encountered in certain industrial effluents, willhydrolyze to some extent even at the pH of 9.5 at which distillation is carriedout.4.2Residual chorine must be removed by pretreatment of the sample with sodiumthiosulfate or other reagents before distillation.4.3Method interferences may be caused by contaminants in the reagent water,reagents, glassware, and other sample processing apparatus that bias analyteresponse.5.0SAFETY5.1The toxicity or carcinogenicity of each reagent used in this method have notbeen fully established. Each chemical should be regarded as a potential healthhazard and exposure should be as low as reasonably achievable. Cautions areincluded for known extremely hazardous materials or procedures.5.2Each laboratory is responsible for maintaining a current awareness file ofOSHA regulations regarding the safe handling of the chemicals specified inthis method. A reference file of Material Safety Data Sheets (MSDS) should bemade available to all personnel involved in the chemical analysis. Thepreparation of a formal safety plan is also advisable.5.3The following chemicals have the potential to be highly toxic or hazardous,consult MSDS.5.3.1Sulfuric acid (Section 7.6)5.3.2Phenol (Section 7.7)5.3.3Sodium nitroprusside (Section 7.10)6.0EQUIPMENT AND SUPPLIES6.1Balance - Analytical, capable of accurately weighing to the nearest 0.0001 g.6.2Glassware - Class A volumetric flasks and pipets as required.6.3An all-glass distilling apparatus with an 800-1000 mL flask.6.4Automated continuous flow analysis equipment designed to deliver and reactsample and reagents in the required order and ratios.6.4.1Sampling device (sampler)6.4.2Multichannel pump350.1-46.4.3Reaction unit or manifold6.4.4Colorimetric detector6.4.5Data recording device7.0REAGENTS AND STANDARDS7.1Reagent water - Ammonia free: Such water is best prepared by passagethrough an ion exchange column containing a strongly acidic cation exchangeresin mixed with a strongly basic anion exchange resin. Regeneration of thecolumn should be carried out according to the manufacturer's instructions.Note: All solutions must be made with ammonia-free water.7.2Boric acid solution (20 g/L): Dissolve 20 g H BO (CASRN 10043-35-3) in33reagent water and dilute to 1 L.7.3Borate buffer: Add 88 mL of 0.1 N NaOH (CASRN 1310-73-2) solution to 500mL of 0.025 M sodium tetraborate solution (5.0 g anhydrous Na B O [CASRN2471330-43-4] or 9.5 g Na B O10H O [CASRN 1303-96-4] per L) and dilute to 1 L2472with reagent water.7.4Sodium hydroxide, 1 N: Dissolve 40 g NaOH in reagent water and dilute to 1L.7.5Dechlorinating reagents: A number of dechlorinating reagents may be used toremove residual chlorine prior to distillation. These include:7.5.1Sodium thiosulfate: Dissolve 3.5 g Na S O5H O (CASRN 10102-17-7)2232in reagent water and dilute to 1 L. One mL of this solution willremove 1 mg/L of residual chlorine in 500 mL of sample.7.5.2Sodium sulfite: Dissolve 0.9 g Na2SO (CASRN 7757-83-7) in reagent3water and dilute to 1 L. One mL removes 1 mg/L Cl per 500 mL ofsample.7.6Sulfuric acid 5 N: Air scrubber solution. Carefully add 139 mL of conc.sulfuric acid (CASRN 7664-93-9) to approximately 500 mL of reagent water.Cool to room temperature and dilute to 1 L with reagent water.7.7Sodium phenolate: Using a 1-L Erlenmeyer flask, dissolve 83 g phenol(CASRN 108-95-2) in 500 mL of distilled water. In small increments,cautiously add with agitation, 32 g of NaOH. Periodically cool flask underwater faucet. When cool, dilute to 1 L with reagent water.7.8Sodium hypochlorite solution: Dilute 250 mL of a bleach solution containing5.25% NaOCl (CASRN 7681-52-9) (such as "Clorox") to 500 mL with reagent350.1-5water. Available chlorine level should approximate 2-3%. Since "Clorox" is aproprietary product, its formulation is subject to change. The analyst mustremain alert to detecting any variation in this product significant to its use inthis procedure. Due to the instability of this product, storage over an extendedperiod should be avoided.7.9Disodium ethylenediamine-tetraacetate (EDTA) (5%): Dissolve 50 g of EDTA(disodium salt) (CASRN 6381-92-6) and approximately six pellets of NaOH in 1L of reagent water.7.10Sodium nitroprusside (0.05%): Dissolve 0.5 g of sodium nitroprusside (CASRN14402-89-2) in 1 L of reagent water.7.11Stock solution: Dissolve 3.819 g of anhydrous ammonium chloride, NH Cl4 (CASRN 12125-02-9), dried at 105°C, in reagent water, and dilute to 1 L.1.0 mL = 1.0 mg NH-N.37.12Standard Solution A: Dilute 10.0 mL of stock solution (Section 7.11) to 1 Lwith reagent water. 1.0 mL = 0.01 mg NH-N.37.13Standard Solution B: Dilute 10.0 mL of standard solution A (Section 7.12) to100.0 mL with reagent water. 1.0 mL = 0.001 mg NH-N.38.0SAMPLE COLLECTION, PRESERVATION AND STORAGE8.1Samples should be collected in plastic or glass bottles. All bottles must bethoroughly cleaned and rinsed with reagent water. Volume collected should besufficient to insure a representative sample, allow for replicate analysis (ifrequired), and minimize waste disposal.8.2Samples must be preserved with H SO to a pH <2 and cooled to 4°C at the24time of collection.8.3Samples should be analyzed as soon as possible after collection. If storage isrequired, preserved samples are maintained at 4°C and may be held for up to28 days.9.0QUALITY CONTROL9.1Each laboratory using this method is required to operate a formal qualitycontrol (QC) program. The minimum requirements of this program consist ofan initial demonstration of laboratory capability, and the periodic analysis oflaboratory reagent blanks, fortified blanks and other laboratory solutions as acontinuing check on performance. The laboratory is required to maintainperformance records that define the quality of the data that are generated.9.2INITIAL DEMONSTRATION OF PERFORMANCE350.1-6350.1-79.2.1The initial demonstration of performance is used to characterizeinstrument performance (determination of LCRs and analysis of QCS)and laboratory performance (determination of MDLs) prior toperforming analyses by this method.9.2.2Linear Calibration Range (LCR) -- The LCR must be determinedinitially and verified every six months or whenever a significant changein instrument response is observed or expected. The initialdemonstration of linearity must use sufficient standards to insure thatthe resulting curve is linear. The verification of linearity must use aminimum of a blank and three standards. If any verification dataexceeds the initial values by ± 10%, linearity must be reestablished. Ifany portion of the range is shown to be nonlinear, sufficient standardsmust be used to clearly define the nonlinear portion.9.2.3Quality Control Sample (QCS) -- When beginning the use of thismethod, on a quarterly basis or as required to meet data-quality needs,verify the calibration standards and acceptable instrument performancewith the preparation and analyses of a QCS. If the determinedconcentrations are not within ±10% of the stated values, performance ofthe determinative step of the method is unacceptable. The source ofthe problem must be identified and corrected before either proceedingwith the initial determination of MDLs or continuing with on-goinganalyses.9.2.4Method Detection Limit (MDL) -- MDLs must be established for allanalytes, using reagent water (blank) fortified at a concentration of twoto three times the estimated instrument detection limit. To determine9MDL values, take seven replicate aliquots of the fortified reagent waterand process through the entire analytical method. Perform allcalculations defined in the method and report the concentration valuesin the appropriate units. Calculate the MDL as follows:where,t = Student's t value for a 99% confidence level and astandard deviation estimate with n-1 degrees offreedom [t = 3.14 for seven replicates]S = standard deviation of the replicate analyses MDLs should be determined every six months, when a new operatorbegins work or whenever there is a significant change in thebackground or instrument response.9.3ASSESSING LABORATORY PERFORMANCE9.3.1Laboratory Reagent Blank (LRB) -- The laboratory must analyze at leastone LRB with each batch of samples. Data produced are used to assess contamination from the laboratory environment. Values that exceed the MDL indicate laboratory or reagent contamination should be suspectedand corrective actions must be taken before continuing the analysis.9.3.2Laboratory Fortified Blank (LFB) -- The laboratory must analyze at leastone LFB with each batch of samples. Calculate accuracy as percentrecovery (Section 9.4.2). If the recovery of any analyte falls outside therequired control limits of 90-110%, that analyte is judged out of control, and the source of the problem should be identified and resolved beforecontinuing analyses.9.3.3The laboratory must use LFB analyses data to assess laboratoryperformance against the required control limits of 90-110%. Whensufficient internal performance data become available (usually aminimum of 20-30 analyses), optional control limits can be developedfrom the percent mean recovery (x) and the standard deviation (S) ofthe mean recovery. These data can be used to establish the upper andlower control limits as follows:UPPER CONTROL LIMIT = x + 3SLOWER CONTROL LIMIT = x - 3SThe optional control limits must be equal to or better than the requiredcontrol limits of 90-110%. After each five to 10 new recoverymeasurements, new control limits can be calculated using only the most recent 20-30 data points. Also, the standard deviation (S) data shouldbe used to established an on-going precision statement for the level ofconcentrations included in the LFB. These data must be kept on fileand be available for review.9.3.4Instrument Performance Check Solution (IPC) -- For all determinationsthe laboratory must analyze the IPC (a mid-range check standard) anda calibration blank immediately following daily calibration, after every10th sample (or more frequently, if required) and at the end of thesample run. Analysis of the IPC solution and calibration blankimmediately following calibration must verify that the instrument iswithin ±10% of calibration. Subsequent analyses of the IPC solutionmust verify the calibration is still within ±10%. If the calibration cannot be verified within the specified limits, reanalyze the IPC solution. If the second analysis of the IPC solution confirms calibration to be outsidethe limits, sample analysis must be discontinued, the cause determinedand/or in the case of drift, the instrument recalibrated. All samplesfollowing the last acceptable IPC solution must be reanalyzed. Theanalysis data of the calibration blank and IPC solution must be kept onfile with the sample analyses data.350.1-8350.1-99.4ASSESSING ANALYTE RECOVERY AND DATA QUALITY9.4.1Laboratory Fortified Sample Matrix (LFM) -- The laboratory must add aknown amount of analyte to a minimum of 10% of the routine samples.In each case the LFM aliquot must be a duplicate of the aliquot usedfor sample analysis. The analyte concentration must be high enough tobe detected above the original sample and should not be less than fourtimes the MDL. The added analyte concentration should be the sameas that used in the laboratory fortified blank.9.4.2Calculate the percent recovery for each analyte, corrected forconcentrations measured in the unfortified sample, and compare thesevalues to the designated LFM recovery range 90-110%. Percentrecovery may be calculate using the following equation:where,R =percent recoveryC =fortified sample concentrationsC =sample background concentrations =concentration equivalent of analyte added tosample9.4.3If the recovery of any analyte falls outside the designated LFM recoveryrange and the laboratory performance for that analyte is shown to be incontrol (Section 9.3), the recovery problem encountered with the LFM isjudged to be either matrix or solution related, not system related.9.4.4Where reference materials are available, they should be analyzed toprovide additional performance data. The analysis of referencesamples is a valuable tool for demonstrating the ability to perform themethod acceptably.10.0CALIBRATION AND STANDARDIZATION10.1Prepare a series of at least three standards, covering the desired range, and ablank by diluting suitable volumes of standard solutions (Sections 7.12 and7.13) to 100 mL with reagent water.10.2Process standards and blanks as described in Section 11.0, Procedure.10.3Set up manifold as shown in Figure 1.10.4Prepare flow system as described in Section 11.0, Procedure.10.5Place appropriate standards in the sampler in order of decreasingconcentration and perform analysis.10.6Prepare standard curve by plotting instrument response against concentrationvalues. A calibration curve may be fitted to the calibration solutionsconcentration/response data using computer or calculator based regressioncurve fitting techniques. Acceptance or control limits should be establishedusing the difference between the measured value of the calibration solutionand the "true value" concentration.10.7After the calibration has been established, it must be verified by the analysis ofa suitable QCS. If measurements exceed ±10% of the established QCS value,the analysis should be terminated and the instrument recalibrated. The newcalibration must be verified before continuing analysis. Periodic reanalysis ofthe QCS is recommended as a continuing calibration check.11.0PROCEDURE11.1Preparation of equipment: Add 500 mL of reagent water to an 800 mLKjeldahl flask. The addition of boiling chips that have been previously treatedwith dilute NaOH will prevent bumping. Steam out the distillation apparatusuntil the distillate shows no trace of ammonia.11.2Sample preparation: Remove the residual chorine in the sample by addingdechlorinating agent (Section 7.5) equivalent to the chlorine residual. To 400mL of sample add 1 N NaOH (Section 7.4), until the pH is 9.5, check the pHduring addition with a pH meter or by use of a short range pH paper.11.3Distillation: Transfer the sample, the pH of which has been adjusted to 9.5, toan 800 mL Kjeldahl flask and add 25 mL of the borate buffer (Section 7.3).Distill 300 mL at the rate of 6-10 mL/min. into 50 mL of 2% boric acid (Section7.2) contained in a 500 mL Erlenmeyer flask.Note: The condenser tip or an extension of the condenser tip must extendbelow the level of the boric acid solution.11.4Since the intensity of the color used to quantify the concentration is pHdependent, the acid concentration of the wash water and the standardammonia solutions should approximate that of the samples.11.5Allow analysis system to warm up as required. Feed wash water throughsample line.11.6Arrange ammonia standards in sampler in order of decreasing concentration ofnitrogen. Complete loading of sampler tray with unknown samples.11.7Switch sample line from reagent water to sampler and begin analysis.350.1-1012.0DATA ANALYSIS AND CALCULATIONS12.1Prepare a calibration curve by plotting instrument response against standardconcentration. Compute sample concentration by comparing sample responsewith the standard curve. Multiply answer by appropriate dilution factor.12.2Report only those values that fall between the lowest and the highestcalibration standards. Samples exceeding the highest standard should bediluted and reanalyzed.12.3Report results in mg NH-N/L.313.0METHOD PERFORMANCE13.1In a single laboratory (EMSL-Cincinnati), using surface water samples atconcentrations of 1.41, 0.77, 0.59, and 0.43 mg NH-N/L, the standard3deviation was ±0.005.13.2In a single laboratory (EMSL-Cincinnati), using surface water samples atconcentrations of 0.16 and 1.44 mg NH-N/L, recoveries were 107% and 99%,3respectively.13.3The interlaboratory precision and accuracy data in Table 1 were developedusing a reagent water matrix. Values are in mg NH-N/L.314.0POLLUTION PREVENTION14.1Pollution prevention encompasses any technique that reduces or eliminates thequantity or toxicity of waste at the point of generation. Numerousopportunities for pollution prevention exist in laboratory operation. The EPAhas established a preferred hierarchy of environmental management techniquesthat places pollution prevention as the management option of first choice.Whenever feasible, laboratory personnel should use pollution preventiontechniques to address their waste generation. When wastes cannot be feasiblyreduced at the source, the Agency recommends recycling as the next bestoption.14.2The quantity of chemicals purchased should be based on expected usageduring its shelf life and disposal cost of unused material. Actual reagentpreparation volumes should reflect anticipated usage and reagent stability.14.3For information about pollution prevention that may be applicable tolaboratories and research institutions, consult "Less is Better: LaboratoryChemical Management for Waste Reduction", available from the AmericanChemical Society's Department of Government Regulations and Science Policy,1155 16th Street N.W., Washington, D.C. 20036, (202)872-4477.15.0WASTE MANAGEMENT350.1-1115.1The U.S. Environmental Protection Agency requires that laboratory wastemanagement practices be conducted consistent with all applicable rules andregulations. Excess reagents, samples and method process wastes should becharacterized and disposed of in an acceptable manner. The Agency urgeslaboratories to protect the air, water and land by minimizing and controllingall releases from hoods, and bench operations, complying with the letter and spirit of any waste discharge permit and regulations, and by complying with all solid and hazardous waste regulations, particularly the hazardous wasteidentification rules and land disposal restrictions. For further information onwaste management consult the "Waste Management Manual for LaboratoryPersonnel", available from the American Chemical Society at the address listed in Section 14.3.350.1-1216.0REFERENCES1.Hiller, A., and Van Slyke, D., "Determination of Ammonia in Blood", J. Biol.Chem. 102, p. 499 (1933).2.O'Connor, B., Dobbs, R., Villiers, B., and Dean. R., "Laboratory Distillation ofMunicipal Waste Effluents", JWPCF 39, R 25 (1967).3.Fiore, J., and O'Brien, J.E., "Ammonia Determination by Automatic Analysis",Wastes Engineering 33, p. 352 (1962).4. A Wetting Agent Recommended and Supplied by the Technicon Corporationfor Use in AutoAnalyzers.5.ASTM "Manual on Industrial Water and Industrial Waste Water", 2nd Ed.,1966 printing, p. 418.6.Booth, R.L., and Lobring. L.B., "Evaluation of the AutoAnalyzer II: A ProgressReport" in Advances in Automated Analysis: 1972 Technicon InternationalCongress, Vol. 8, p. 7-10, Mediad Incorporated, Tarrytown, N.Y., (1973).7.Standards Methods for the Examination of Water and Wastewater, 18thEdition, p. 4-77, Methods 4500 NH3 B and H (1992).8.Annual Book of ASTM Standards, Part 31, "Water", Standard D1426-79(C).9.Code of Federal Regulations 40, Ch. 1, Pt. 136, Appendix B.350.1-1317.0TABLES, DIAGRAMS, FLOWCHARTS, AND VALIDATION DATATABLE 1. INTERLABORATORY PRECISION AND ACCURACY DATA Number of True StandardValues Value Mean Residual Deviation ResidualReported(T)(X)for X(S)for S 1340.2700.2670-0.00110.03420.00151570.6920.69720.00590.0476-0.0070136 1.20 1.20080.00010.0698-0.0112195 1.60 1.60950.00760.10230.0006142 3.00 3.01280.00690.1677-0.0067159 3.50 3.4991-0.00830.21680.0165156 3.60 3.5955-0.01220.1821-0.0234200 4.20 4.22710.01770.28550.04881968.768.7257-0.05680.4606-0.012715611.011.07470.04570.5401-0.049514213.012.9883-0.04650.69610.002719918.017.9727-0.0765 1.16350.2106 REGRESSIONS: X = 1.003T - 0.003, S = 0.052T + 0.019350.1-14。
epa3500方法

epa3500方法
EPA 3500方法是美国环境保护局(EPA)所制定的一种分析化
学方法,用于土壤、废水、废物和其它环境样品中有机化合物的测定。
该方法通常用于环境监测和污染物检测。
EPA 3500方法主要涉
及样品的制备、提取和分析,以便准确测定有机化合物的含量。
具
体来说,该方法通常包括以下步骤:
1. 样品的准备,样品需要根据方法要求进行适当的处理和准备,以确保分析的准确性和可重复性。
2. 提取过程,样品中的有机化合物需要通过适当的提取方法提
取出来,以便后续的分析。
3. 分析方法,EPA 3500方法通常使用气相色谱-质谱联用
(GC-MS)或液相色谱-质谱联用(LC-MS)等高灵敏度的分析技术,
以确保对有机化合物的准确测定。
4. 质量控制,在整个分析过程中,需要进行质量控制以确保分
析结果的可靠性。
总的来说,EPA 3500方法是一种标准化的分析化学方法,用于环境样品中有机化合物的测定。
通过严格遵循该方法,可以获得准确可靠的分析结果,从而评估环境中有机污染物的水平,为环境保护和管理提供重要的数据支持。
美国国家环保局EPA方法要点和推荐仪器

美国国家环保局EPA方法要点和推荐仪器EPA方法218.6离子色谱测定在饮用水、地下水和工业废水中的水溶性铬(1994年修订版3.3)应用范围测定饮用水、地下水和工业废水中的水溶性六价铬(如CrO2-4),这种方法的检测下限为0.4μg/L。
样品中如果含有大量的阴离子物质如硫酸或氯离子可能会引起色谱柱过载。
样品如果含有大量有机物或硫离子可能会引起可溶性的六价铬快速还原为三价铬。
样品贮存在4℃,在24小时内分析。
方法采用离子色谱法分析。
方法要点:水样经0.45μm滤膜过滤后,用浓缓冲溶液调节pH为9-9.5。
样品的测量体积为50-250μL进样到离子色谱。
保护柱去除样品中的有机物,六价铬以CrO2-4形式,在高容量的阴离子交换分离柱上分离,六价铬用双苯基苄巴脲柱后衍生,然后在530nm波长下检测有色络合物。
建议采用的仪器条件保护柱:Dionex IonPac NG1或与之相同的色谱柱分离柱:Dionex IonPac AS7或与之相同的色谱柱阴离子抑制器装置:Dionex Anion MicroMembrane Suppressor,其它抑制器必须有足够低的检测限和足够的基线稳定性。
色谱条件:色谱柱:保护柱-Dionex IonPac NG1, 分离柱-Dionex IonPac AS7淋洗液:250mM (NH4)2SO4, 100mM NH4OH, 流速=1.5 mL/min柱后试剂:2mM双苯基苄巴脲,10% v/v甲醇,1N 硫酸,流速=0.5 mL/min 检测器:可见光530nm保留时间:3.8 分钟离子色谱测定无机阴离子(1993年八月,修订版2.2)应用范围1.可测定的阴离子包括A部分:溴离子,氯离子,氟离子,硝酸根,亚硝酸根,磷酸根,硫酸B部分:溴酸根,亚氯酸根,氯酸根2.基体包括:饮用水,地表水,民用水和工业废水,地下水,试剂用水,固体浸出液方法要点1.小量样品,一般2-3mL注入离子色谱,阴离子采用一个系统含有保护柱,分离柱,抑制器和电导检测器进行分离和检测。
美国EPA通用土壤筛选值

75-68-3
1,1-二氟-1-氯乙烷
5.8E+04
ns
2.4E+05
nms
5.2E+01
1.0E+05
n
126-99-8
2-氯-1,3-丁二烯
8.4E+00
n
3.6E+01
n
7.5E-03
1.4E+01
n
3165-93-3
1.5E+02
n
1.5E+03
n
4.7E+01
9.1E+01
n
7664-41-7
氨
7790-98-9
高氯酸铵
5.5E+01
n
7.2E+02
n
2.6E+01
n
7773-06Leabharlann 0氨基磺酸铵1.6E+04
n
2.0E+05
nm
7.3E+03
n
62-53-3
苯胺
8.5E+01
c**
3.0E+02
c*
4.0E-03
c
542-88-1
二氯甲基醚
7.7E-05
c
3.9E-04
c
1.5E-08
6.2E-05
c
80-05-7
双酚A
3.1E+03
n
3.1E+04
n
1.4E+02
1.8E+03
n
7440-42-8
硼及硼酸盐
1.6E+04
n
2.0E+05
美国EPA 关于空气自动监测系统性能指标的规定和测试方法
美国EPA关于大气自动监测系统性能指标的规定和测试方法引言环境空气污染的自动监测方法有多种,一般采用湿法和干法两种。
湿法是基于化学量理论的库仑法和电导法等测量原理,需使用大量试剂,存在试剂调整和废液处理等问题,操作比较繁琐,故障率较高,维护工作量较大;干法是基于物理光谱测量理论,使样品始终保持在气体状态,没有试剂的损耗,维护工作量较小。
比如SO2测量采用紫外荧光法,NOx测量采用化学发光法,O3测量采用紫外光度法,CO测量采用气体过滤相关分析法等,目前我国绝大部分空气自动监测采用的是该方法。
干法测量以欧美为主。
美国开展空气自动监测已有30年的历史,在空气自动监测方面积累了丰富的经验,并制定了详细的规范。
其中物理光谱法作为美国EPA的推荐方法,得到了广泛的应用。
湿法测量以日本为主,但自1996年起日本在法定的测量方法中增加了干式测量法。
利用物质的光谱特性进行污染物的分析已成为自动监测仪器发展的必然趋势。
我国在环境空气质量监测和质量保证方面的规定都参考了美国国家环保署(EPA)的规定。
目前,大气自动监测和空气质量日报工作在我国大部分省市已广泛开展,自动监测仪器监测数据的准确可靠是日报工作中的基础。
为使监测人员了解美国EPA关于空气自动监测的相关规定,特将其有关SO2、NO2、O3、CO自动监测仪器的性能指标规定和测试方法作简要说明,以供参考。
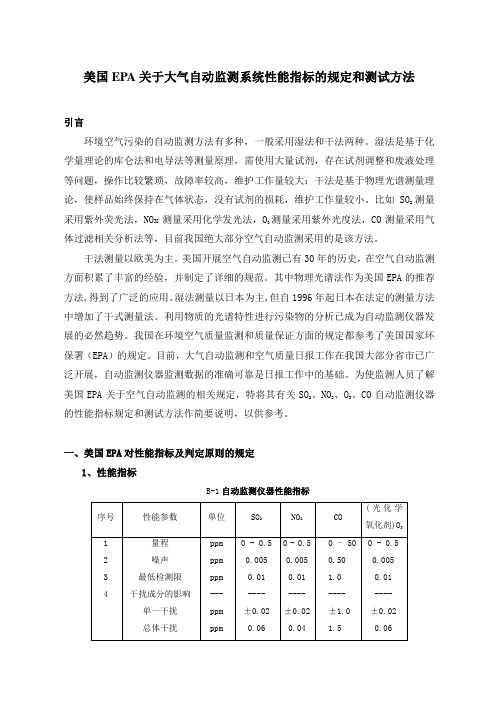
一、美国EPA对性能指标及判定原则的规定1、性能指标B-1自动监测仪器性能指标M/0.02447,M是该气体的摩尔质量。
2、判定原则对于每个性能指标(量程除外),测试程序从开始起要重复7次,得到7组测试结果。
每组结果要和表B-1中的规定指标相比较,高于或超出规定指标的值是一个超标值。
每个参数的7个结果说明如下:(1)0次超标:被测的参数合格;(2)3次或更多次超标:该参数不合格;(3)1次或2次超标:再重复测试该参数 8次,得到共15个测试结果。
将此15个测试结果说明如下:a:1次或2次超标:通过测试;b:3次以上:该参数不合格。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
METHOD 3520CCONTINUOUS LIQUID-LIQUID EXTRACTION1.0SCOPE AND APPLICATION1.1This method describes a procedure for isolating organic compounds from aqueous samples. The method also describes concentration techniques suitable for preparing the extract for the appropriate determinative steps described in Sec. 4.3 of Chapter Four.1.2This method is applicable to the isolation and concentration of water-insoluble and slightly soluble organics in preparation for a variety of chromatographic procedures.1.3Method 3520 is designed for extraction solvents with greater density than the sample. Continuous extraction devices are available for extraction solvents that are less dense than the sample. The analyst must demonstrate the effectiveness of any such automatic extraction device before employing it in sample extraction.1.4This method is restricted to use by or under the supervision of trained analysts. Each analyst must demonstrate the ability to generate acceptable results with this method.2.0SUMMARY OF METHOD2.1 A measured volume of sample, usually 1 liter, is placed into a continuous liquid-liquid extractor, adjusted, if necessary, to a specific pH (see Table 1), and extracted with organic solvent for 18 - 24 hours.2.2The extract is dried, concentrated (if necessary), and, as necessary, exchanged into a solvent compatible with the cleanup or determinative method being employed (see Table 1 for appropriate exchange solvents).3.0INTERFERENCES3.1Refer to Method 3500.3.2The decomposition of some analytes has been demonstrated under basic extraction conditions required to separate analytes. Organochlorine pesticides may dechlorinate, phthalate esters may exchange, and phenols may react to form tannates. These reactions increase with increasing pH, and are decreased by the shorter reaction times available in Method 3510. Method 3510 is preferred over Method 3520 for the analysis of these classes of compounds. However, the recovery of phenols may be optimized by using Method 3520 and performing the initial extraction at the acid pH.4.0APPARATUS AND MATERIALS4.1Continuous liquid-liquid extractor - Equipped with polytetrafluoroethylene (PTFE) or glass connecting joints and stopcocks requiring no lubrication (Kontes 584200-0000, 584500-0000, 583250-0000, or equivalent).CD-ROM3520C - 1Revision 3December 19964.2Drying column - 20 mm ID Pyrex® chromatographic column with Pyrex® glass wool at bottom and a PTFE stopcock.NOTE:Fritted glass discs are difficult to decontaminate after highly contaminated extracts have been passed through. Columns without frits may be purchased.Use a small pad of Pyrex® glass wool to retain the adsorbent. Prewash theglass wool pad with 50 mL of acetone followed by 50 mL of elution solvent priorto packing the column with adsorbent.4.3Kuderna-Danish (K-D) apparatus4.3.1Concentrator tube - 10-mL graduated (Kontes K-570050-1025 or equivalent).A ground glass stopper is used to prevent evaporation of extracts.4.3.2Evaporation flask - 500-mL (Kontes K-570001-500 or equivalent). Attach toconcentrator tube with springs, clamps, or equivalent.4.3.3Snyder column - Three-ball macro (Kontes K-503000-0121 or equivalent).4.3.4Snyder column - Two-ball micro (Kontes K-569001-0219 or equivalent).4.3.5Springs - 1/2 inch (Kontes K-662750 or equivalent).NOTE:The following glassware is recommended for the purpose of solvent recovery during the concentration procedures requiring the use of Kuderna-Danishevaporative concentrators. Incorporation of this apparatus may be requiredby State or local municipality regulations that govern air emissions of volatileorganics. EPA recommends the incorporation of this type of reclamationsystem as a method to implement an emissions reduction program. Solventrecovery is a means to conform with waste minimization and pollutionprevention initiatives.4.4Solvent vapor recovery system (Kontes K-545000-1006 or K-547300-0000, Ace Glass 6614-30, or equivalent).4.5Boiling chips - Solvent-extracted, approximately 10/40 mesh (silicon carbide or equivalent).4.6Water bath - Heated, with concentric ring cover, capable of temperature control (± 5E C). The bath should be used in a hood.4.7Vials - 2-mL, glass with PTFE-lined screw-caps or crimp tops.4.8pH indicator paper - pH range including the desired extraction pH.4.9Heating mantle - Rheostat controlled.4.10Syringe - 5-mL.CD-ROM3520C - 2Revision 3December 1996CD-ROM 3520C - 3Revision 3December 19965.0REAGENTS5.1Reagent grade chemicals shall be used in all tests. Unless otherwise indicated, it isintended that all reagents shall conform to the specifications of the Committee on AnalyticalReagents of the American Chemical Society, where such specifications are available. Other gradesmay be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit itsuse without lessening the accuracy of the determination. Reagents should be stored in glass toprevent the leaching of contaminants from plastic containers.5.2Organic-free reagent water - All references to water in this method refer to organic-freereagent water, as defined in Chapter One.5.3Sodium hydroxide solution (10N), NaOH. Dissolve 40 g NaOH in organic-free reagentwater and dilute to 100 mL. Other concentrations of hydroxide solutions may be used to adjustsample pH, provided that the volume added does not appreciably change (e.g., <1%) the totalsample volume.5.4Sodium sulfate (granular, anhydrous), Na SO . Purify by heating at 400E C for 4 hours24in a shallow tray, or by precleaning the sodium sulfate with methylene chloride. If the sodium sulfateis precleaned with methylene chloride, a method blank must be analyzed, demonstrating that thereis no interference from the sodium sulfate.5.5Sulfuric acid solution (1:1 v/v), H SO . Slowly add 50 mL of H SO (sp. gr. 1.84) to 5024 24mL of organic-free reagent water. Other concentrations of acid solutions may be used to adjustsample pH, provided that the volume added does not appreciably change (e.g., <1%) the totalsample volume.5.6Extraction/exchange solvents - All solvents must be pesticide quality or equivalent.5.6.1Methylene chloride, CH Cl , boiling point 39E C. 225.6.2Hexane, C H , boiling point 68.7E C.6145.6.32-Propanol, CH CH(OH)CH , boiling point 82.3E C. 335.6.4Cyclohexane, C H , boiling point 80.7E C. 6125.6.5Acetonitrile, CH CN, boiling point 81.6E C.36.0SAMPLE COLLECTION, PRESERVATION, AND HANDLINGSee the introductory material to this chapter, Organic Analytes, Sec. 4.1.7.0PROCEDURE7.1Using a 1-liter graduated cylinder, measure 1 liter (nominal) of sample. Alternatively, ifthe entire contents of sample bottle are to be extracted, mark the level of sample on the outside ofthe bottle. If high concentrations are anticipated, a smaller sample volume may be taken and dilutedto 1-L with organic-free reagent water. It is recommended that if high analyte concentrations areanticipated, samples should be collected in smaller sample bottles and the whole sample used.7.2Pipet 1.0 mL of the surrogate spiking solution into each sample in the graduated cylinder (or sample bottle) and mix well. (See Method 3500 and the determinative method to be used for details on the surrogate standard solution and matrix spiking solution).7.2.1For the sample in each batch (see Chapter One) selected for use as a matrixspike sample, add 1.0 mL of the matrix spiking standard.7.2.2If Method 3640, Gel-Permeation Cleanup, is to be employed, add twice thevolume of the surrogate spiking solution and the matrix spiking standard, since half of the extract is not recovered from the GPC apparatus. (Alternatively, use 1.0 mL of the spiking solutions and concentrate the final extract to half the normal volume, e.g., 0.5 mL instead of1.0 mL).7.3Check the pH of the sample with wide-range pH paper and adjust the pH, if necessary, to the pH indicated in Table 1, using 1:1 (v/v) sulfuric acid or 10 N sodium hydroxide. Lower concentrations of acid or base solution may be employed, provided that they do not result in a significant change (<1%) in the volume of sample extracted (see Secs. 5.3 and 5.5).7.4Add 300 - 500 mL of methylene chloride to the distilling flask of the extractor. Add several boiling chips to the flask.7.5Quantitatively transfer the sample from the graduated cylinder (or sample bottle) to the extractor. Use a small volume of methylene chloride to rinse the cylinder (or bottle) and transfer this rinse solvent to the extractor. Add organic-free reagent water to the extractor, if needed, to ensure proper operation and extract for 18-24 hours. If the sample was transferred directly from the sample bottle, refill the bottle to the mark made in Sec. 7.1 with water and then measure the volume of sample that was in the bottle.7.6Allow the extractor to cool, then detach the boiling flask. If extraction at a secondary pH is not required (see Table 1), the extract is dried and concentrated using one of the techniques described in Secs. 7.10 - 7.11.7.7If a pH adjustment and second extraction is required (see Table 1), carefully, while stirring, adjust the pH of the aqueous phase to the second pH indicated in Table 1. If the extracts are to be analyzed separately (see Sec. 7.8), attach a clean distilling flask containing 500 mL of methylene chloride to the continuous extractor. Extract for 18-24 hours, allow to cool, and detach the distilling flask. If the extracts are not to be analyzed separately, then the distilling flask and solvent need not be changed and may be used for the second pH extraction.7.8If performing GC/MS analysis (Method 8270), the acid/neutral and base extracts may be combined prior to concentration. However, in some situations, separate concentration and analysis of the acid/neutral and base extracts may be preferable (e.g., if for regulatory purposes the presence or absence of specific acid/neutral and base compounds at low concentrations must be determined, separate extract analyses may be warranted).7.9Perform concentration (if necessary) using the Kuderna-Danish technique (Secs. 7.10.1 through 7.10.6).7.10K-D technique7.10.1Assemble a Kuderna-Danish (K-D) concentrator (Sec. 4.3) by attaching a 10-mLconcentrator tube to a 500-mL evaporation flask.CD-ROM3520C - 4Revision 3December 19967.10.2Attach the solvent vapor recovery glassware (condenser and collection device)(Sec. 4.4) to the Snyder column of the K-D apparatus following manufacturer's instructions.7.10.3Dry the extract by passing it through a drying column containing about 10 cm ofanhydrous sodium sulfate. Collect the dried extract in a K-D concentrator. Rinse the Erlenmeyer flask, which contained the solvent extract, with 20 - 30 mL of methylene chloride and add it to the column to complete the quantitative transfer.7.10.4Add one or two clean boiling chips to the flask and attach a three-ball Snydercolumn. Prewet the Snyder column by adding about 1 mL of methylene chloride to the top of the column. Place the K-D apparatus on a hot water bath (15 - 20E C above the boiling point of the solvent) so that the concentrator tube is partially immersed in the hot water and the entire lower rounded surface of the flask is bathed with hot vapor. Adjust the vertical position of the apparatus and the water temperature, as required, to complete the concentration in 10 -20 minutes. At the proper rate of distillation the balls of the column will actively chatter, butthe chambers will not flood. When the apparent volume of liquid reaches 1 mL, remove the K-D apparatus from the water bath and allow it to drain and cool for at least 10 minutes.7.10.5If a solvent exchange is required (as indicated in Table 1), momentarily removethe Snyder column, add 50 mL of the exchange solvent, a new boiling chip, and reattach the Snyder column. Alternatively, pour the exchange solvent into the top of the Snyder column while the concentrator remains on the water bath in Sec. 7.10.4. Concentrate the extract, as described in Sec. 7.10.4, raising the temperature of the water bath, if necessary, to maintain proper distillation.7.10.6Remove the Snyder column and rinse the flask and its lower joints into theconcentrator tube with 1 - 2 mL of methylene chloride or exchange solvent. If sulfur crystals are a problem, proceed to Method 3660 for cleanup. The extract may be further concentrated by using the techniques outlined in Sec. 7.11 or adjusted to 10.0 mL with the solvent last used.7.11If further concentration is indicated in Table 1, either the micro-Snyder column technique (7.11.1) or nitrogen blowdown technique (7.11.2) is used to adjust the extract to the final volume required.7.11.1Micro-Snyder column techniqueAdd another one or two clean boiling chips to the concentrator tube and attacha two-ball micro-Snyder column. Prewet the column by adding 0.5 mL of methylenechloride or exchange solvent to the top of the column. Place the K-D apparatus in a hotwater bath so that the concentrator tube is partially immersed in the hot water. Adjustthe vertical position of the apparatus and the water temperature, as required, to completethe concentration in 5 - 10 minutes. At the proper rate of distillation the balls of thecolumn will actively chatter, but the chambers will not flood. When the apparent volumeof liquid reaches 0.5 mL, remove the K-D apparatus from the water bath and allow it todrain and cool for at least 10 minutes. Remove the Snyder column, rinse the flask andits lower joints into the concentrator tube with 0.2 mL of methylene chloride or theexchange solvent, and adjust the final volume as indicated in Table 1, with solvent. CD-ROM3520C - 5Revision 3December 19967.11.2Nitrogen blowdown technique7.11.2.1Place the concentrator tube in a warm bath (35E C) and evaporate thesolvent to the final volume indicated in Table 1, using a gentle stream of clean, drynitrogen (filtered through a column of activated carbon).CAUTION:New plastic tubing must not be used between the carbon trap andthe sample, since it may introduce contaminants.7.11.2.2The internal wall of the tube must be rinsed several times withmethylene chloride or appropriate solvent during the operation. During evaporation, thetube must be positioned to avoid water condensation (i.e., the solvent level should bebelow the level of the water bath). Under normal procedures, the extract must not beallowed to become dry.CAUTION:When the volume of solvent is reduced below 1 mL, semivolatileanalytes may be lost.7.12The extract may now be analyzed for the target analytes using the appropriate determinative technique(s) (see Sec. 4.3 of this chapter). If analysis of the extract will not be performed immediately, stopper the concentrator tube and store refrigerated. If the extract will be stored longer than 2 days it should be transferred to a vial with a PTFE-lined screw-cap or crimp top, and labeled appropriately.8.0QUALITY CONTROL8.1Any reagent blanks, matrix spikes, or replicate samples should be subjected to exactly the same analytical procedures as those used on actual samples.8.2Refer to Chapter One for specific quality control procedures and Method 3500 for extraction and sample-preparation procedures.9.0METHOD PERFORMANCERefer to the determinative methods for performance data.10.0REFERENCESNone.CD-ROM3520C - 6Revision 3December 1996CD-ROM 3520C - 8Revision 3December 1996METHOD 3520CCONTINUOUS LIQUID-LIQUID EXTRACTION。