美国环保局 EPA 试验 方法 8318
美国EPA快速评价体系

美国EPA快速评价体系
美国EPA快速评价体系:EPA是美国环境保护署(U.SEnvironmentalProtectionAgency)的英文缩写。
美国EPA快速评价体系的主要任务是保护人类健康和自然环境。
EPA总部设在华盛顿, 有 10 个地方办公室和几十个实验室。
在全美国有18000 名雇员。
他们半数以上是工程师 , 科学家和政策分析家。
负责对很多环境项目设立国家标准 , 监控强制性标准的执行和符合情况。
EPA 联合州和地方政府颁发一系列商业以及工业许可证。
EPA认证美国环保总署EPA的主要目的是保护人民健康、保护生态环境--空气、水和土地这些我们赖以生存的环境。
在成立之后的30多年,EPA一直在为给全美人民创造一个整洁的健康环境而努力。
如果符合EPA要求,则EPA会颁发符合证书。
epa毒理参数和筛选值

epa毒理参数和筛选值一、概述EPA(美国环保署)毒理参数和筛选值是用于评估化学品毒性的重要标准。
这些参数和值是根据大量的科学研究和实践经验得出的,用于指导化学品的安全使用和管理。
本文将介绍EPA毒理参数和筛选值的基本概念、目的和用途。
二、定义与范围EPA毒理参数和筛选值通常涉及化学品的生物累积性、毒性效应、暴露评估等方面。
这些参数和值适用于各种环境介质(如水、空气、土壤等)和生物体,包括人类和其他动物。
这些参数和值的范围广泛,包括急性毒性、慢性毒性、生态毒性、致畸毒性等。
三、评估方法评估化学品毒性通常采用实验方法,包括动物实验和人体研究。
实验过程中,需要确定合适的剂量范围和暴露时间,以模拟实际环境中的暴露情况。
实验结果将用于计算化学品对生物体的毒性效应,并据此得出相应的毒理参数和筛选值。
四、毒理参数与筛选值的差异毒理参数是指化学品对生物体造成危害的综合能力,通常由一组实验结果得出。
而筛选值是指针对特定目标(如特定组织或器官)或特定生物群体的化学品毒性参数的较低阈值,用于初步判断化学品是否可能对生物体造成危害。
五、应用与影响EPA毒理参数和筛选值对于环境保护和公共健康至关重要。
它们为化学品的风险评估和管理提供了依据,有助于制定合理的政策和管理措施,确保公共安全和生态环境不受损害。
此外,这些参数和值也为科研人员提供了研究化学品毒性的基础数据,有助于推动毒理学研究的发展。
六、结论EPA毒理参数和筛选值是评估化学品毒性的重要标准,涵盖了广泛的化学品和环境介质。
通过实验方法和科学研究,这些参数和值被用来评估化学品的综合毒性,并确定较低的阈值用于初步判断化学品是否可能对生物体造成危害。
这些参数和值对于环境保护和公共健康至关重要,为化学品的风险评估和管理提供了依据,有助于制定合理的政策和管理措施,确保公共安全和生态环境不受损害。
七、建议与展望为了更好地应对化学品对环境和人类健康的威胁,建议加强毒理学研究,提高毒理参数和筛选值的准确性和适用性。
美国EPA最新参考方法标准

07/25/06
环境 S.A 公司
Graseby Andersen/GMW 公司
MP101M 型 PM10 监测 器
1200 型大容量空气采样 器
自动等价方法: 配置 40CFR 50 附录 L 中特定
EQPM-0404-151 的带通风孔的 PM10 进气口或
其先前平顶式版本及三个可选
联邦公告:卷 69, 温控采样管(RST),操作满量
RFPS-1287-063 1200 型 PM10 特定粒径进口和
标识为 SAUV-10H、SAUV-11H、
联邦公报:卷 52, GMW-IP-10、GMW-IP-10-70、
第 45684 页 , GMW-IP-10-801
或
12/01/87 及卷 53, GMW-IP-10-8000 中的任何一个
联邦公报:卷 63, 孔的进口,作为 PM10 参考方法 第 69625 页 , 配置,流量为 16.67 升/分钟,24
12/17/98
小时连续采样周期操作。原有固
最 新 修 订 : 件版本为 5.X 和稍低或者新固
PQ100 操作说明书或修订版
06/23/99
/分钟,24 小时连续采样周期操
作。符合 RAAS105-200 操作说
明书,遵循 40 CFR 第 50 部分,
附录 J 或附录 M 中有关要求和
特别规定的样品采集过滤器。
手动参考方法: 配备 RAAS-10 PM10 进口或附
RFPS-0699-132 录 L,40 联邦法规(CFR)50,
第 1062 页 , 大容量采样器,这些采样器含有
01/15/88
以下部件:带有丙烯腈-丁二烯-
苯乙烯塑胶过滤器托架和电机/
EPA方法索引

EPA方法索引和相关标准品EPA 是美国国家环境保护局(U.S Environmental Protection Agency) 的英文缩写。
它的主要任务是保护人类健康和自然环境。
EPA 制定了一系列标准分析方法用于环境监测领域。
主要包括:EPA T01~T14 系列标准分析方法——空气中有毒有机物分析方法EPA IP1~IP10 系列标准分析方法——室内空气污染物的分析测定方法EPA 200 系列标准分析方法———金属的分析方法EPA 500 系列标准分析方法——饮用水中有机物的分析方法EPA 600 系列标准分析方法——城市和工业废水中有机化合物的分析方法SW -846 系列标准分析方法——固体废弃物试验分析评价手册1300 系列是毒性试验方法3000 系列是金属元素的提取方法3500 系列是半(非) 挥发性有机物的提取方法3600 系列是净化、分离方法5000 系列是挥发性有机物的提取方法6000 系列是测定金属的新方法7000 系列是原子吸收法测定金属元素8000 系列是有机物分析方法9000 系列是常规项目分析方法其中,500系列,600系列和8000系列是环境种有机物分析最常用的方法。
EPA 600系列方法是美国为贯彻“净水法”(CW A) 、“全国水体污染物排放消除制度”(NPDES) 和“许可证制度”,严格控制点源排放,保护地表水,使其免受城市和工业废水中有机物的污染而制定的。
EPA 500 系列方法是为执行“安全饮用水法”(SDW A) 和“国家一级饮用水法案”(National Primary Drinking Water Regulations) ,确保饮用水及饮用水源的质量而制订的。
EPA 500 系列是针对比较洁净的水样(饮用水、地下水、地表水) 开发的,有些方法仅用试剂水和饮用水验证过SW-846 系列集中贯彻了“资源保护回收法”和“陆地处置限制法规”的精神,包含了固体废弃物采样和分析试验的全部方法, 是在EPA200 ~EPA 600 系列的基础上发展起来的。
美国EPA排放法规
美国高速公路摩托车排放法规介绍尹涛(天津大学天津摩托车技术中心)Yin Tao (Tianjin University Tianjin Motorcycle Technical center)高速公路摩托车排放法规自1980年生效20多年来未做过修订,2002年7月,美国环保署提出了重新修订高速公路摩托车排放法规的议案,2005年正式推出了法规的修订本,其法规排放试验限值变化如表1所示。
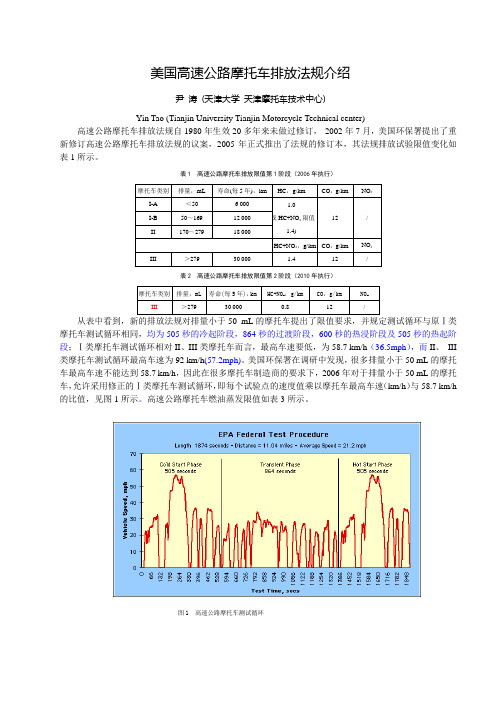
表1 高速公路摩托车排放限值第1阶段(2006年执行)表2 高速公路摩托车排放限值第2阶段(2010年执行)从表中看到,新的排放法规对排量小于50 mL的摩托车提出了限值要求,并规定测试循环与原Ⅰ类摩托车测试循环相同,均为505秒的冷起阶段,864秒的过渡阶段,600秒的热浸阶段及505秒的热起阶段;Ⅰ类摩托车测试循环相对II、III类摩托车而言,最高车速要低,为58.7 km/h(36.5mph),而II、III 类摩托车测试循环最高车速为92 km/h(57.2mph)。
美国环保署在调研中发现,很多排量小于50 mL的摩托车最高车速不能达到58.7 km/h,因此在很多摩托车制造商的要求下,2006年对于排量小于50 mL的摩托车,允许采用修正的Ⅰ类摩托车测试循环,即每个试验点的速度值乘以摩托车最高车速(km/h)与58.7 km/h 的比值,见图1所示。
高速公路摩托车燃油蒸发限值如表3所示。
图1 高速公路摩托车测试循环表3 EPA燃油蒸发标准(2008年执行)以证明高速公路摩托车符合蒸发排放标准,二是通过设计声明40 CFR 1051.245(e)来证明可以达到蒸发排放标准要求。
试验流程图如图2所示,设计声明格式如表4所示。
表4 设计声明符合标准*备注:根据耐久试验情况可以缩短静置时间全部试验流程利用劣化系数的试验流程图2 试验流程(收稿日期200505-09)。
EPA测试方法的检索

EPA测试方法的检索EPA(环境保护局)是美国的主要环境保护机构,负责制定和推动环境保护的相关政策和规定。
为了保证环境的质量和可持续性,EPA开发了许多测试方法,用于评估和监测环境中的污染物和污染源。
下面是一些可以帮助您检索EPA测试方法的方法和资源。
2.EPA指南和手册:EPA还发布了大量的指南、手册和技术文件,介绍了各种测试方法,以及如何使用和实施这些方法。
这些指南和手册通常包含了测试方法的具体步骤、仪器和设备的要求、样品采集和处理的指导等信息。
您可以在EPA的官方网站上相关指南和手册,或者浏览EPA的出版物目录以获取相关资源。
3. EPA在线数据库:EPA还提供了一些在线数据库,可以帮助您查找和访问EPA测试方法和相关资料。
其中最著名的是Test Methods for Evaluating Solid Waste - Physical/Chemical Methods (SW-846)。
这本手册收集了用于评估固体废物的物理化学方法的详细信息。
您可以在EPA的官方网站或其他相关资源中找到SW-846手册的最新版本和相关补充材料。
除了上述方法和资源外,还有一些其他途径可以检索EPA测试方法。
您可以参考相关领域的学术期刊和文献,了解最新的环境测试方法和技术。
此外,许多大学和研究机构也在开展与环境测试相关的研究,并且发布相关的文献和方法。
通过参与相关的学术研究项目或参加学术会议,您可以了解到更多的测试方法和新兴的环境监测技术。
需要注意的是,EPA测试方法的选择和使用应根据具体的需求和应用来进行。
不同国家和地区在环境监测和评估方面有不同的要求和标准,因此您还需要根据当地的法规和政策进行相关调查和分析。
在实施测试方法之前,还应了解所需方法的准确性、可靠性和适用性,并确保操作人员具备必要的技术能力和实施经验。
美国EPA 关于空气自动监测系统性能指标的规定和测试方法
美国EPA关于大气自动监测系统性能指标的规定和测试方法引言环境空气污染的自动监测方法有多种,一般采用湿法和干法两种。
湿法是基于化学量理论的库仑法和电导法等测量原理,需使用大量试剂,存在试剂调整和废液处理等问题,操作比较繁琐,故障率较高,维护工作量较大;干法是基于物理光谱测量理论,使样品始终保持在气体状态,没有试剂的损耗,维护工作量较小。
比如SO2测量采用紫外荧光法,NOx测量采用化学发光法,O3测量采用紫外光度法,CO测量采用气体过滤相关分析法等,目前我国绝大部分空气自动监测采用的是该方法。
干法测量以欧美为主。
美国开展空气自动监测已有30年的历史,在空气自动监测方面积累了丰富的经验,并制定了详细的规范。
其中物理光谱法作为美国EPA的推荐方法,得到了广泛的应用。
湿法测量以日本为主,但自1996年起日本在法定的测量方法中增加了干式测量法。
利用物质的光谱特性进行污染物的分析已成为自动监测仪器发展的必然趋势。
我国在环境空气质量监测和质量保证方面的规定都参考了美国国家环保署(EPA)的规定。
目前,大气自动监测和空气质量日报工作在我国大部分省市已广泛开展,自动监测仪器监测数据的准确可靠是日报工作中的基础。
为使监测人员了解美国EPA关于空气自动监测的相关规定,特将其有关SO2、NO2、O3、CO自动监测仪器的性能指标规定和测试方法作简要说明,以供参考。
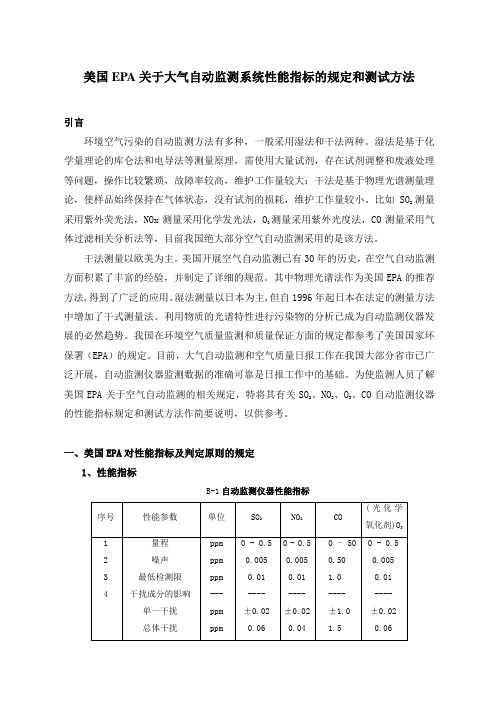
一、美国EPA对性能指标及判定原则的规定1、性能指标B-1自动监测仪器性能指标M/0.02447,M是该气体的摩尔质量。
2、判定原则对于每个性能指标(量程除外),测试程序从开始起要重复7次,得到7组测试结果。
每组结果要和表B-1中的规定指标相比较,高于或超出规定指标的值是一个超标值。
每个参数的7个结果说明如下:(1)0次超标:被测的参数合格;(2)3次或更多次超标:该参数不合格;(3)1次或2次超标:再重复测试该参数 8次,得到共15个测试结果。
将此15个测试结果说明如下:a:1次或2次超标:通过测试;b:3次以上:该参数不合格。
epa测试标准

epa测试标准
EPA(美国环保局)的测试标准是针对各种污染物和产品制定的,旨在确保产品符合相关法规和标准,以保护环境和人类健康。
以下是一些EPA的测试标准:
污染物排放限制:EPA制定了一系列污染物排放限制,要求企业、工厂和机构遵守。
这些限制涵盖了空气、水和土壤中的污染物,如二氧化硫、氮氧化物、挥发性有机化合物等。
消费品安全标准:EPA负责制定和执行消费品安全标准,以确保消费品不会对人类健康造成危害。
这些标准涵盖了家电、家具、儿童用品、玩具、化妆品等产品。
农药注册与评价:EPA负责评价农药的安全性和有效性,以确保农药的使用不会对人类健康和环境造成危害。
化学物质管理:EPA负责管理和限制化学物质的生产和使用,以确保这些物质不会对环境和人类健康造成危害。
环保设备与系统性能测试:EPA还制定了各种环保设备与系统性能测试标准,以确保这些设备能够有效地降低污染物的排放。
以上是一些EPA的测试标准,具体标准可能因产品、污染物和法规而有所不同。
在进行相关测试时,建议咨询专业的环保机构或实验室,以确保测试结果的准确性和可靠性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
METHOD 8318N-METHYLCARBAMATES BY HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC)1.0SCOPE AND APPLICATION1.1Method 8318 is used to determine the concentration of N-methylcarbamates in soil, water and waste matrices. The following compounds can be determined by this method:_______________________________________________________________________________Compound Name CAS No.a________________________________________________________________________________Aldicarb (Temik) 116-06-3Aldicarb Sulfone 1646-88-4Carbaryl (Sevin) 63-25-2Carbofuran (Furadan) 1563-66-2Dioxacarb 6988-21-23-Hydroxycarbofuran16655-82-6Methiocarb (Mesurol) 2032-65-7Methomyl (Lannate)16752-77-5Promecarb 2631-37-0Propoxur (Baygon) 114-26-1________________________________________________________________________________aChemical Abstract Services Registry Number.1.2The method detection limits (MDLs) of Method 8318 for determining the target analytes in organic-free reagent water and in soil are listed in Table 1.1.3This method is restricted to use by, or under the supervision of, analysts experienced in the use of high performance liquid chromatography (HPLC) and skilled in the interpretation of chromatograms. Each analyst must demonstrate the ability to generate acceptable results with this method.2.0SUMMARY OF METHOD2.1N-methylcarbamates are extracted from aqueous samples with methylene chloride, and from soils, oily solid waste and oils with acetonitrile. The extract solvent is exchanged to methanol/ethylene glycol, and then the extract is cleaned up on a C-18 cartridge, filtered, and eluted on a C-18 analytical column. After separation, the target analytes are hydrolyzed and derivatized post-column, then quantitated fluorometrically.2.2Due to the specific nature of this analysis, confirmation by a secondary method is not essential. However, fluorescence due to post-column derivatization may be confirmed by substituting the NaOH and o-phthalaldehyde solutions with organic-free reagent water and reanalyzing the sample. Iffluorescence is still detected, then a positive interference is present and care should be taken in the interpretation of the results.2.3The sensitivity of the method usually depends on the level of interferences present, rather than on the instrumental conditions. Waste samples with a high level of extractable fluorescent compounds are expected to yield significantly higher detection limits.3.0INTERFERENCES3.1Fluorescent compounds, primarily alkyl amines and compounds which yield primary alkyl amines on base hydrolysis, are potential sources of interferences.3.2Coeluting compounds that are fluorescence quenchers may result in negative interferences.3.3Impurities in solvents and reagents are additional sources of interferences. Before processing any samples, the analyst must demonstrate daily, through the analysis of solvent blanks, that the entire analytical system is interference free.4.0APPARATUS AND MATERIALS4.1HPLC system4.1.1An HPLC system capable of injecting 20 µL aliquots andperforming multilinear gradients at a constant flow. The system must also be equipped with a data system to measure the peak areas.4.1.2C-18 reverse phase HPLC column, 25 cm x 4.6 mm (5 µm).4.1.3Post Column Reactor with two solvent delivery systems (KratosPCRS 520 with two Kratos Spectroflow 400 Solvent Delivery Systems, or equivalent).4.1.4Fluorescence detector (Kratos Spectroflow 980, or equivalent).4.2Other apparatus4.2.1Centrifuge.4.2.2Analytical balance - + 0.0001 g.4.2.3Top loading balance - + 0.01 g.4.2.4Platform shaker.4.2.5Heating block, or equivalent apparatus, that can accommodate10 mL graduated vials (Sec. 4.3.11).4.3Materials4.3.1HPLC injection syringe - 50 µL.4.3.2Filter paper, (Whatman #113 or #114, or equivalent).4.3.3Volumetric pipettes, Class A, glass, assorted sizes.R4.3.4Reverse phase cartridges, (C-18 Sep-Pak [Waters Associates],or equivalent).4.3.5Glass syringes - 5 mL.4.3.6Volumetric flasks, Class A - Sizes as appropriate.4.3.7Erlenmeyer flasks with teflon-lined screw caps, 250 mL.4.3.8Assorted glass funnels.4.3.9Separatory funnels, with ground glass stoppers and teflonstopcocks - 250 mL.4.3.10Graduated cylinders - 100 mL.4.3.11Graduated glass vials - 10 mL, 20 mL.4.3.12Centrifuge tubes - 250 mL.4.3.13Vials - 25 mL, glass with Teflon lined screw caps orcrimp tops.4.3.14Positive displacement micro-pipettor, 3 to 25 µLdisplacement, (Gilson Microman [Rainin #M-25] with tips, [Rainin #CP-25], or equivalent).4.3.15Nylon filter unit, 25 mm diameter, 0.45 µm pore size,disposable (Alltech Associates, #2047, or equivalent).5.0REAGENTS5.1HPLC grade chemicals shall be used in all tests. It is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Other grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lowering the accuracy of the determination.5.2General5.2.1Acetonitrile, CH CN - HPLC grade - minimum UV cutoff at 203 nm3(EM Omnisolv #AX0142-1, or equivalent).5.2.2Methanol, CH OH - HPLC grade - minimum UV cutoff at 230 nm (EM3Omnisolv #MX0488-1, or equivalent).5.2.3Methylene chloride, CH Cl - HPLC grade - minimum UV cutoff at22230 nm (EM Omnisolv #DX0831-1, or equivalent).5.2.4Hexane, C H - pesticide grade - (EM Omnisolv #HX0298-1, or614equivalent).5.2.5Ethylene glycol, HOCH CH OH - Reagent grade - (EM Science, or22equivalent).5.2.6Organic-free reagent water - All references to water in this method refer to organic-free reagent water, as defined in Chapter One.5.2.7Sodium hydroxide, NaOH - reagent grade - 0.05N NaOH solution.5.2.8Phosphoric acid, H PO - reagent grade.345.2.9pH 10 borate buffer (J.T. Baker #5609-1, or equivalent).5.2.10o-Phthalaldehyde, o-C H(CHO) - reagent grade (Fisher642#0-4241, or equivalent).5.2.112-Mercaptoethanol, HSCH CH OH - reagent grade (Fisher22#0-3446, or equivalent).5.2.12N-methylcarbamate neat standards (equivalence to EPA standards must be demonstrated for purchased solutions).5.2.13Chloroacetic acid, ClCH COOH, 0.1 N.25.3Reaction solution5.3.1Dissolve 0.500 g of o-phthalaldehyde in 10 mL of methanol, ina 1 L volumetric flask. To this solution, add 900 mL of organic-free reagent water, followed by 50 mL of the borate buffer (pH 10). After mixing well, add 1 mL of 2-mercaptoethanol, and dilute to the mark with organic-free reagent water. Mix the solution thoroughly. Prepare fresh solutions on a weekly basis, as needed. Protect from light and store under refrigeration.5.4Standard solutions5.4.1Stock standard solutions: prepare individual 1000 mg/L solutions by adding 0.025 g of carbamate to a 25 mL volumetric flask, and diluting to the mark with methanol. Store solutions, under refrigeration, in glass vials with Teflon lined screw caps or crimp tops. Replace every six months.5.4.2Intermediate standard solution: prepare a mixed 50.0 mg/L solution by adding 2.5 mL of each stock solution to a 50 mL volumetric flask, and diluting to the mark with methanol. Store solutions, underrefrigeration, in glass vials with Teflon lined screw caps or crimp tops.Replace every three months.5.4.3Working standard solutions: prepare 0.5, 1.0, 2.0, 3.0 and 5.0mg/L solutions by adding 0.25, 0.5, 1.0, 1.5 and 2.5 mL of the intermediate mixed standard to respective 25 mL volumetric flasks, and diluting each to the mark with methanol. Store solutions, under refrigeration, in glass vials with Teflon lined screw caps or crimp tops.Replace every two months, or sooner if necessary.5.4.4Mixed QC standard solution: prepare a 40.0 mg/L solution fromanother set of stock standard solutions, prepared similarly to those described in Sec. 5.4.1. Add 2.0 mL of each stock solution to a 50 mL volumetric flask and dilute to the mark with methanol. Store the solution, under refrigeration, in a glass vial with a Teflon lined screw cap or crimp top. Replace every three months.6.0SAMPLE COLLECTION, PRESERVATION, AND HANDLING6.1Due to the extreme instability of N-methylcarbamates in alkaline media, water, waste water and leachates should be preserved immediately after collection by acidifying to pH 4-5 with 0.1 N chloroacetic acid.6.2Store samples at 4E C and out of direct sunlight, from the time of collection through analysis. N-methylcarbamates are sensitive to alkaline hydrolysis and heat.6.3All samples must be extracted within seven days of collection, and analyzed within 40 days of extraction.7.0PROCEDURE7.1Extraction7.1.1Water, domestic wastewater, aqueous industrial wastes, andleachates7.1.1.1Measure 100 mL of sample into a 250 mL separatoryfunnel and extract by shaking vigorously for about 2 minutes with 30mL of methylene chloride. Repeat the extraction two more times.Combine all three extracts in a 100 mL volumetric flask and diluteto volume with methylene chloride. If cleanup is required, go toSec. 7.2. If cleanup is not required, proceed directly to Sec.7.3.1.7.1.2Soils, solids, sludges, and heavy aqueous suspensions7.1.2.1Determination of sample % dry weight - In certaincases, sample results are desired based on dry-weight basis. Whensuch data is desired, a portion of sample for this determinationshould be weighed out at the same time as the portion used for analytical determination.WARNING:The drying oven should be contained in a hood orvented. Significant laboratory contamination mayresult from a heavily contaminated hazardouswaste sample.7.1.2.1.1Immediately after weighing the sample forextraction, weigh 5-10 g of the sample into a tared crucible.Determine the % dry weight of the sample by drying overnightoat 105C. Allow to cool in a desiccator before weighing:% dry weight = g of dry sample x 100g of sample7.1.2.2Extraction - Weigh out 20 + 0.1 g of sample intoa 250 mL Erlenmeyer flask with a Teflon-lined screw cap. Add 50 mLof acetonitrile and shake for 2 hours on a platform shaker. Allow the mixture to settle (5-10 min), then decant the extract into a 250 mL centrifuge tube. Repeat the extraction two more times with 20 mL of acetonitrile and 1 hour shaking each time. Decant and combine all three extracts. Centrifuge the combined extract at 200 rpm for10 min. Carefully decant the supernatant into a 100 mL volumetricflask and dilute to volume with acetonitrile. (Dilution factor = 5) Proceed to Sec. 7.3.2.7.1.3Soils heavily contaminated with non-aqueous substances, such as oils7.1.3.1Determination of sample % dry weight - FollowSecs. 7.1.2.1 through 7.1.2.1.1.7.1.3.2Extraction - Weigh out 20 + 0.1 g of sample intoa 250 mL Erlenmeyer flask with a Teflon-lined screw cap. Add 60 mLof hexane and shake for 1 hour on a platform shaker. Add 50 mL of acetonitrile and shake for an additional 3 hours. Allow the mixture to settle (5-10 min), then decant the solvent layers into a 250 mL separatory funnel. Drain the acetonitrile (bottom layer) through filter paper into a 100 mL volumetric flask. Add 60 mL of hexane and50 mL of acetonitrile to the sample extraction flask and shake for1 hour. Allow the mixture to settle, then decant the mixture intothe separatory funnel containing the hexane from the first extraction. Shake the separatory funnel for 2 minutes, allow the phases to separate, drain the acetonitrile layer through filter paper into the volumetric flask, and dilute to volume with acetonitrile. (Dilution factor = 5) Proceed to Sec. 7.3.2.7.1.4Non-aqueous liquids such as oils7.1.4.1Extraction - Weigh out 20 + 0.1 g of sample intoa 125 mL separatory funnel. Add 40 mL of hexane and 25 mL ofacetonitrile and vigorously shake the sample mixture for 2 minutes.Allow the phases to separate, then drain the acetonitrile (bottomlayer) into a 100 mL volumetric flask. Add 25 mL of acetonitrile tothe sample funnel, shake for 2 minutes, allow the phases toRepeat the extraction with another 25 mL portion of acetonitrile,combining the extracts. Dilute to volume with acetonitrile.(Dilution factor = 5). Proceed to Sec. 7.3.2.7.2Cleanup - Pipet 20.0 mL of the extract into a 20 mL glass vial containing 100 µL of ethylene glycol. Place the vial in a heating block set at o50 C, and gently evaporate the extract under a stream of nitrogen (in a fume hood) until only the ethylene glycol keeper remains. Dissolve the ethylene glycol residue in 2 mL of methanol, pass the extract through a pre-washed C-18 reverse phase cartridge, and collect the eluate in a 5 mL volumetric flask. Elute the cartridge with methanol, and collect the eluate until the final volume of 5.0 mL is obtained. (Dilution factor = 0.25) Using a disposable 0.45 µm filter, filter an aliquot of the clean extract directly into a properly labelled autosampler vial. The extract is now ready for analysis. Proceed to Sec. 7.4.7.3Solvent Exchange7.3.1Water, domestic wastewater, aqueous industrial wastes, andleachates:Pipet 10.0 mL of the extract into a 10 mL graduated glass vial containing 100 µL of ethylene glycol. Place the vial in a heating block oset at 50 C, and gently evaporate the extract under a stream of nitrogen (in a fume hood) until only the ethylene glycol keeper remains. Add methanol to the ethylene glycol residue, dropwise, until the total volume is 1.0 mL. (Dilution factor = 0.1). Using a disposable 0.45 µm filter, filter this extract directly into a properly labelled autosampler vial.The extract is now ready for analysis. Proceed to Sec. 7.4.7.3.2Soils, solids, sludges, heavy aqueous suspensions, and non-aqueous liquids:Elute 15 mL of the acetonitrile extract through a C-18 reverse phase cartridge, prewashed with 5 mL of acetonitrile. Discard the first 2 mL of eluate and collect the remainder. Pipet 10.0 mL of the clean extract intoa 10 mL graduated glass vial containing 100 µL of ethylene glycol. Placeothe vial in a heating block set at 50 C, and gently evaporate the extract under a stream of nitrogen (in a fume hood) until only the ethylene glycol keeper remains. Add methanol to the ethylene glycol residue, dropwise, until the total volume is 1.0 mL. (Additional dilution factor = 0.1;overall dilution factor = 0.5). Using a disposable 0.45 µm filter, filter this extract directly into a properly labelled autosampler vial. The extract is now ready for analysis. Proceed to Sec. 7.4.7.4Sample Analysis7.4.1Analyze the samples using the chromatographic conditions,post-column reaction parameters and instrument parameters given in Secs.7.4.1.1, 7.4.1.2, 7.4.1.3 and 7.4.1.4. Table 2 provides the retentiontimes that were obtained under these conditions during method development.A chromatogram of the separation is shown in Figure 1.7.4.1.1Chromatographic Conditions (Recommended)Solvent A:Organic-free reagent water, acidified with0.4 mL of phosphoric acid per liter ofwaterSolvent B:Methanol/acetonitrile (1:1, v/v)Flow rate: 1.0 mL/minInjection Volume:20 µLSolvent delivery system program:Time Duration(min)Function Value(min) File0.00 FR 1.0 00.00 B% 10% 00.02 B% 80% 20 020.02 B%100% 5 025.02 B%100% 5 030.02 B% 10% 3 033.02 B% 10% 7 036.02 ALARM 0.01 07.4.1.2Post-column Hydrolysis Parameters (Recommended)Solution:0.05 N aqueous sodium hydroxideFlow Rate:0.7 mL/minoTemperature 95 CResidence Time:35 seconds (1 mL reaction coil)7.4.1.3Post-column Derivatization Parameters(Recommended)Solution:o-phthalaldehyde/2-mercaptoethanol (Sec.5.3.1)Flow Rate:0.7 mL/minoTemperature 40 CResidence time:25 seconds (1 mL reaction coil)7.4.1.4Fluorometer Parameters (Recommended)Cell:10 µLExcitation wavelength:340 nmEmission waveleng 418 nm cutoff filterSensitivity wavelength:0.5 µAPMT voltage: -800 VTime constant: 2 sec7.4.2If the peak areas of the sample signals exceed the calibration range of the system, dilute the extract as necessary and reanalyze the diluted extract.7.5Calibration:7.5.1Analyze a solvent blank (20 µL of methanol) to ensure that thesystem is clean. Analyze the calibration standards (Sec. 5.4.3), starting with the 0.5 mg/L standards and ending with the 5.0 mg/L standard. If the percent relative standard deviation (%RSD) of the mean response factor (RF) for each analyte does not exceed 20%, the system is calibrated and the analysis of samples may proceed. If the %RSD for any analyte exceeds 20%, recheck the system and/or recalibrate with freshly prepared calibration solutions.7.5.2Using the established calibration mean response factors, checkthe calibration of the instrument at the beginning of each day by analyzing the 2.0 mg/L mixed standard. If the concentration of each analyte falls within the range of 1.70 to 2.30 mg/L (i.e., within + 15% of the true value), the instrument is considered to be calibrated and the analysis of samples may proceed. If the observed value of any analyte exceeds its true value by more than + 15%, the instrument must be recalibrated (Sec. 7.5.1).7.5.3After 10 sample runs, or less, the 2.0 mg/L standards must beanalyzed to ensure that the retention times and response factors are still within acceptable limits. Significant variations (i.e., observed concentrations exceeding the true concentrations by more than + 15%) may require a re-analysis of the samples.7.6Calculations7.6.1Calculate each response factor as follows (mean value based on5 points):concentration of standard RF =area of the signal5(3 RF )i i mean RF = RF =__ 55[(3 RF - RF)] / 4i __21/2i %RSD of RF = X 100%__RF __7.6.2Calculate the concentration of each N-methylcarbamate asfollows:µg/g or mg/L = (RF) (area of signal) (dilution factor)__8.0QUALITY CONTROL8.1Before processing any samples, the analyst must demonstrate, through the analysis of a method blank for each matrix type, that all glassware and reagents are interference free. Each time there is a change of reagents, a method blank must be processed as a safeguard against laboratory contamination.8.2 A QC check solution must be prepared and analyzed with each sample batch that is processed. Prepare this solution, at a concentration of 2.0 mg/L of each analyte, from the 40.0 mg/L mixed QC standard solution (Sec. 5.4.4). The acceptable response range is 1.7 to 2.3 mg/L for each analyte.8.3Negative interference due to quenching may be examined by spiking the extract with the appropriate standard, at an appropriate concentration, and examining the observed response against the expected response.8.4Confirm any detected analytes by substituting the NaOH and OPA reagents in the post column reaction system with deionized water, and reanalyze the suspected extract. Continued fluorescence response will indicate that a positive interference is present (since the fluorescence response is not due to the post column derivatization). Exercise caution in the interpretation of the chromatogram.9.0METHOD PERFORMANCE9.1Table 1 lists the single operator method detection limit (MDL) for each compound in organic-free reagent water and soil. Seven/ten replicate samples were analyzed, as indicated in the table. See reference 7 for more details.9.2Tables 2, 3 and 4 list the single operator average recoveries and standard deviations for organic-free reagent water, wastewater and soil. Ten replicate samples were analyzed at each indicated spike concentration for each matrix type.9.3The method detection limit, accuracy and precision obtained will be determined by the sample matrix.10.0REFERENCES1.California Department of Health Services, Hazardous Materials Laboratory,"N-Methylcarbamates by HPLC", Revision No. 1.0, September 14, 1989.2.Krause, R.T. Journal of Chromatographic Science, 1978, vol. 16, pg 281.3.Klotter, Kevin, and Robert Cunico, "HPLC Post Column Detection ofCarbamate Pesticides", Varian Instrument Group, Walnut Creek, CA 94598.EPA, "Method 531. Measurement of N-Methylcarbomyloximes and N-Methylcarbamates in Drinking Water by Direct Aqueous Injection HPLC withPost Column Derivatization", EPA 600/4-85-054, Environmental Monitoring and Support Laboratory, Cincinnati, OH 45268.EPA, "Method 632. The Determination of Carbamate and Urea Pesticides inIndustrial and Municipal Wastewater", EPA 600/4-21-014, Environmental Monitoring and Support Laboratory, Cincinnati, OH 45268.6.Federal Register, "Appendix B to Part 136 - Definition and Procedure forthe Determination of the Method Detection Limit - Revision 1.11", Friday, October 26, 1984, 49, No. 209, 198-199.7.Okamoto, H.S., D. Wijekoon, C. Esperanza, J. Cheng, S. Park, J. Garcha, S.Gill, K. Perera "Analysis for N-Methylcarbamate Pesticides by HPLC in Environmental Samples", Proceedings of the Fifth Annual USEPA Symposium on Waste Testing and Quality Assurance, July 24-28, 1989, Vol. II, 57-71.aELUTION ORDER, RETENTION TIMES ANDSINGLE OPERATOR METHOD DETECTION LIMITSMethod Detection Limits b Compound Retention Organic-freeTime Reagent Water Soil(min) (µg/L)(µg/kg)_______________________________________________________________________________c cAldicarb Sulfone 9.59 1.944Methomyl (Lannate) 9.59 1.7123-Hydroxycarbofuran12.70 2.610cDioxacarb13.50 2.2 >50cc cAldicarb (Temik)16.05 9.412Propoxur (Baygon)18.06 2.417Carbofuran (Furadan)18.28 2.022Carbaryl (Sevin)19.13 1.731d-Naphthol20.30 - -Methiocarb (Mesurol)22.56 3.132Promecarb23.02 2.517________________________________________________________________________________ aSee Sec. 7.4 for chromatographic conditionsbMDL for organic-free reagent water, sand, soil were determined by analyzing 10 low concentration spike replicate for each matrix type (except where noted). See reference 7 for more details.cMDL determined by analyzing 7 spiked replicates.dBreakdown product of Carbaryl.aPRECISION DATA FOR ORGANIC-FREE REAGENT WATERCompound Recovered% Recovery SD%RSD_____________________________________________________________________________ Aldicarb Sulfone22575.0 7.28 3.24 Methomyl (Lannate)24481.3 8.34 3.423-Hydroxycarbofuran21070.0 7.85 3.74 Dioxacarb24180.3 8.53 3.54 Aldicarb (Temik)22474.7 13.5 6.03 Propoxur (Baygon)23277.3 10.6 4.57 Carbofuran (Furadan)23979.6 9.23 3.86 Carbaryl (Sevin)24280.7 8.56 3.54 Methiocarb (Mesurol)23177.0 8.09 3.50 Promecarb22775.7 9.43 4.1_______________________________________________________________________________ aSpike Concentration = 300 µg/L of each compound, n = 10aPRECISION DATA FOR WASTEWATERCompound Recovered% Recovery SD%RSD______________________________________________________________________________Aldicarb Sulfone23578.317.67.49 Methomyl (Lannate)24782.329.912.10 3-Hydroxycarbofuran25183.725.410.11bDioxacarb - - - Aldicarb (Temik)25886.016.4 6.36 Propoxur (Baygon)26387.716.7 6.47 Carbofuran (Furadan)26287.315.7 5.99 Carbaryl (Sevin)26287.317.2 6.56 Methiocarb (Mesurol)25484.719.97.83 Promecarb26387.715.1 5.74 ________________________________________________________________________________ aSpike Concentration = 300 µg/L of each compound, n = 10bNo recoveryaPRECISION DATA FOR SOILCompound Recovered% Recovery SD%RSD______________________________________________________________________________Aldicarb Sulfone 1.5778.50.069 4.39 Methomyl (Lannate) 1.4874.00.086 5.81 3-Hydroxycarbofuran 1.6080.00.071 4.44 Dioxacarb 1.5175.50.073 4.83 Aldicarb (Temik) 1.2964.50.14211.0 Propoxur (Baygon) 1.3366.50.1269.47 Carbofuran (Furadan) 1.4673.00.092 6.30 Carbaryl (Sevin) 1.5376.50.076 4.90 Methiocarb (Mesurol) 1.4572.50.071 4.90 Promecarb 1.2964.70.1249.61 _______________________________________________________________________________ aSpike Concentration = 2.00 mg/kg of each compound, n = 101.00 µg/mL EACH OF:1.ALDICARB SULFONE 6.PROPOXUR2.METHOMYL7.CARBOFURAN3.3-HYDROXYCARBOFURAN8.CARBARYL4.DIOXACARB9.METHIOCARB5.ALDICARB10.PROMECARB。