紫外-2
2紫外吸收光谱分析
紫外吸收光谱分析一概述紫外可见吸收光谱法是利用某些物质的分子吸收10~800nm光谱区的辐射来进行分析测定的方法,这种分子吸收光谱产生于价电子和分子轨道上的电子在电子能级间的跃迁,广泛用于有机和无机物质的定性和定量测定。
该方法具有灵敏度高、准确度好、选择性优操作简便、分析速度好等特点。
分子的紫外可见吸收光谱法是基于分子内电子跃迁产生的吸收光谱进行分析的一种常用的光谱分析法。
分子在紫外-可见区的吸收与其电子结构紧密相关。
紫外光谱的研究对象大多是具有共轭双键结构的分子。
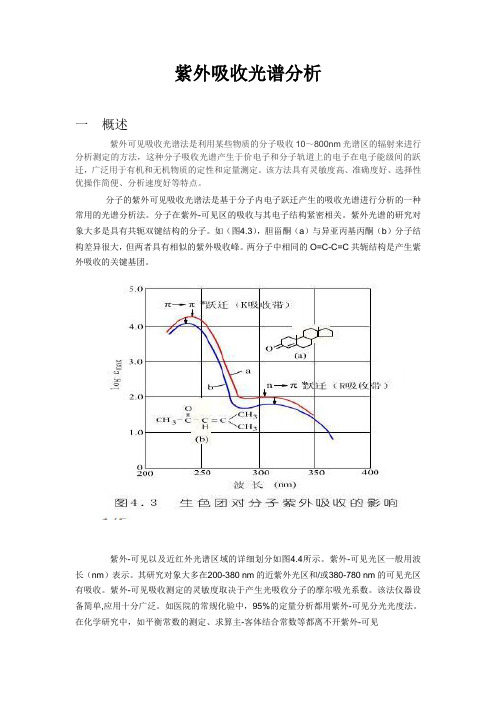
如(图4.3),胆甾酮(a)与异亚丙基丙酮(b)分子结构差异很大,但两者具有相似的紫外吸收峰。
两分子中相同的O=C-C=C共轭结构是产生紫外吸收的关键基团。
紫外-可见以及近红外光谱区域的详细划分如图4.4所示。
紫外-可见光区一般用波长(nm)表示。
其研究对象大多在200-380 nm的近紫外光区和/或380-780 nm的可见光区有吸收。
紫外-可见吸收测定的灵敏度取决于产生光吸收分子的摩尔吸光系数。
该法仪器设备简单,应用十分广泛。
如医院的常规化验中,95%的定量分析都用紫外-可见分光光度法。
在化学研究中,如平衡常数的测定、求算主-客体结合常数等都离不开紫外-可见二基本原理紫外可见吸收光谱的基本原理是利用在光的照射下待测样品内部的电子跃迁,电子跃迁类型有:(1)σ→σ* 跃迁指处于成键轨道上的σ电子吸收光子后被激发跃迁到σ*反键轨道(2)n→σ* 跃迁指分子中处于非键轨道上的n电子吸收能量后向σ*反键轨道的跃迁(3)π→π* 跃迁指不饱和键中的π电子吸收光波能量后跃迁到π*反键轨道。
(4)n→π* 跃迁指分子中处于非键轨道上的n电子吸收能量后向π*反键轨道的跃迁。
电子跃迁类型不同,实际跃迁需要的能量不同:σ→σ* ~150nmn→σ* ~200nmπ→π* ~200nmn→π* ~300nm吸收能量的次序为:σ→σ*>n→σ*≥π→π*>n→π*特殊的结构就会有特殊的电子跃迁,对应着不同的能量(波长),反反映在紫外可见吸收光谱图上就有一定位置一定强度的吸收峰,根据吸收峰的位置和强度就可以推知待测样品的结构信息三特点1、紫外可见吸收光谱所对应的电磁波长较短,能量大,它反映了分子中价电子能级跃迁情况。
2-2紫外光谱

2.2紫外光谱紫外光谱(Ultraviolet Spectroscopy,缩写为UV)是电子吸收光谱,通常所说的紫外光谱的波长范围是200~400nm,常用的紫外光谱仪的测试范围可扩展到可见光区域,包括400~800nm的波长区域。
电子光谱是指分子外层价电子能级跃迁形成的光谱。
用紫外光照射样品时,当样品分子或原子吸收光子后,外层电子由基态跃迁到激发态,不同结构的样品分子,其电子的跃迁方式是不同的,而且吸收光的波长范围不同,吸光的几率也不同,从而可根据波长范围、吸光度鉴别不同物质结构方面的差异。
2.2.1 基本概念2.2.1.1 分子光谱的产生在分子中,除了电子相对于原子核的运动外,还有核间相对位移引起的振动和转动。
这三种运动能量都是量子化的,并对应有一定能级,图2-1为分子的能级示意图。
图2-1 分子中电子能级、振动能级和转动能级示意图在每一电子能级上有许多间距较小的振动能级,在每一振动能级上又有许多更小的转动能级。
若用△E电子、△E振动、△E转动分别表示电子能级、振动能级和转动能级差,即有△E电子△E振动△E转动。
处在同一电子能级的分子,可能因其振动能量不同,而处在不同的振动能级上。
当分子处在同一电子能级和同一振动能级时,它的能量还会因转动能量不同,而处在不同的转动能级上。
所以分子的总能量可以认为是这三种能量的总和:E分子=E电子+E振动+E转动。
当用频率为v的电磁波照射分子,而该分子的较高能级与较低能级之差△E恰好等于该电磁波的能量hv时,即有△E=hv(h为普朗克常数)(2-1)此时,在微观上出现分子由较低的能级跃迁到较高的能级;在宏观上则透射光的强度变小。
若用一连续辐射的电磁波照射分子,将照射前后光强度的变化转变为电信号,并记录下来,然后以波长为横坐标,以电信号(吸光度A)为纵坐标,就可以得到一张光强度变化对波长的关系曲线图—分子吸收光谱图。
分子的转动能级差一般在0.005 ~ 0.05eV。
罗丹明B显色检测Fenton反应产生的羟自由基

化缓慢 ,可见本方法稳定性较好 。 2. 3 工作曲线 当保持其它条件不变 ,改变 RB 的浓度 ,测得它 与体系吸光度的关系。实验表明 RB 的浓度在 0~ 40 μ molΠ L 范围内与吸光度呈良好的线性关系。 工作曲线的线性回归方程为 A = 0. 2329 cRB
2 ( 105 molΠ L ) + 0. 0182 , 线性相关系数 R = 019988 。
(pH 2152) ,用二次蒸馏水稀释到 10 mL 并摇匀 , 放
molΠ L) :称
取 0. 0239 g 罗丹明 B ( 四乙基罗丹明 ) 用水定容 2+ - 3 250 mL容 量 瓶 ; Fe 溶 液 ( 5 ×10 molΠ L) : 称取
013421 g FeSO4 ・ 7H2 O , 加 0. 6 mL0. 5 molΠ L 的稀 H2 SO4 溶解 , 用水定容 250 mL 容量瓶 ; 苯甲酸溶液
LIU Jie and SONG Gong2wu ( The Analysis and Test
Center , Hubei University , Wuhan 430062 ) , Fenxi Shiyanshi ,2004 ,23 ( 10) :55~57 Abstract :A new method was proposed for the determi2 nation of hydroxyl radical produced by Fenton reaction. After being oxidized by hydroxyl radical , Rhodamine B showed a color change. The ΔA 550 was dependent on the dosage of Rhodamine B , Fe
紫外-可见分光光度法 (2)全

2.1 紫外-可见吸收光谱 2.2 紫外-可见光度计仪器组成 2.3 吸收光谱的测量-----Lambert-Beer 定律(新增) 2.4 分析条件选择(新增) 2.5 UV-Vis分光光度法的应用
的或者说将产生非连续谱。因此,分子的能量变化E为各种形式能量变
化的总和:
ΔΕ ΔΕe ΔΕv ΔΕr
其中Ee最大:1-20 eV; Ev次之:0.05-1 eV; Er最小:0.05 eV
可见,电子能级间隔比振动能级和转动能级间隔大1~2个数量级, 在发生电子能级跃迁时,伴有振-转能级的跃迁,形成所谓的带状光谱。
用于盛放样品。可用石英或玻璃两种材料制作,前者适于紫外区和 可见光区;后者只适于可见光区。有些透明有机玻璃亦可用作吸收池。
四、检测器:硒光电池、PMT、PDA
二、紫外可见光度计仪器 分光光度计分为单波
长和双波长仪器。
1. 单波长分光光度计 (a) 单光束 (b) 双光束(空间分隔) (c) 双光束(时间分隔)
如CH2=CH2的-*跃迁,max=165~200nm;而1,3-丁二烯, max=217nm 2)异构现象:使异构物光谱出现差异。
如 CH3CHO 含 水 化 合 物 有 两 种 可 能 的 结 构 : CH3CHO-H2O 及 CH3CH(OH)2; 已烷中,max=290nm,表明有醛基存在,结构为前
不同物质结构不同或者说 其分子能级的能量(各种能级 能量总和)或能量间隔各异, 因此不同物质将选择性地吸收 不同波长或能量的外来辐射, 这是UV-Vis定性分析的基础。
定性分析具体做法是让不 同波长的光通过待测物,经待 测物吸收后,测量其对不同波 长 光 的 吸 收 程 度 ( 吸 光 度 A) , 以吸光度A为纵坐标,辐射波 长为横坐标作图,得到该物质 的吸收光谱或吸收曲线,据吸 收曲线的特性(峰强度、位置 及数目等)研究分子结构。
紫外分光光度法测定维生素B1片含量以及方法验证-2

紫外分光光度法测定维生素B1片含量0845051089 以药学一班及陈秋娟方法证验紫外分光光度法测定维生素B 1片含量以及方法验证班级:08级药学一班 姓名:陈秋娟 学号:0845051089 [摘要]目的:采用紫外分光光度法对维生素B 1片含量进行测定及此方法的验证 方法:运用紫外分光光度法在λ=246nm 处测定吸光度,并运用百分吸收系数(1%1cm E )法(标示量(%)=A ×D ×W 平均×1%1cm E ﹣¹××W ﹣¹×标示量﹣¹×100%和对照品比较法进行计算。
结果:该维生素B 1片维生素B 1含量:用百分吸收系数(1%1cm E )法计算标示量(%)为93.05%;对照品比较法计算标示量(%)为94.33%;用标准曲线法计算标示量为102.87%。
实验方法验证:维生素B 1片含量为7.5μg/ml ~ 17.5μg/ml 时,维生素B 1浓度与吸光度成良好的线性关系;平均回收率(%)为103.53%;RSD(%)为1.17%。
结论:紫外分光光度法对本实验测定专属性好,并且方法简单易操作。
该维生素B 1片维生素B 1含量为93.05%,符合本品含维生素B 1 (C 12H 17ClN 4OS.HCl )应为标示量的90.0%~ 110.0%的规定为合格。
维生素B 1 紫外分光光度法 含量测定 [实验内容] (一)1、仪器与试药电子天平(XX 公司,编号:XX ),XX 紫外分光光度计(编号:XX ,厂家:XX )。
维生素B1(批号:XX ,生产厂家:XX 公司),维生素B1对照品(精制原料,由中国药品生物制品检定所提供,批号:xx ), 辅料空白(根据维生素B1片处方工艺配制),盐酸溶液(9→1000),纯化水 。
2、处方分析:组成 处方量(g)比例(%) 维生素B 1 10 16.39 淀粉 20 32.78 糊精 30 49.18 硬脂酸镁 1.0 1.639 生产1000片根据处方量分析,维生素B 1含16.39%,辅料含83.61%。
环糊精及衍生物_药物包合常数的测定方法及其应用

[接受日期] 20032112123 通讯作者: 任勇,副教授;研究方向: 药物超分子;T el :025********* E 2m ail :2002reny ong @环糊精及衍生物Π药物包合常数的测定方法及其应用王亚娜1, 孙俊梅2, 余丽丽1, 陆亚鹏1, 任 勇13(11南京师范大学新药研究中心,江苏南京210097;21湖北三峡大学医学院,湖北宜昌443001)[摘 要] 包合常数是决定环糊精包合性质的重要参数。
综述环糊精及其衍生物与药物包合物的包合常数的测定方法及其应用,简介近年国内报道的各种β2C D 及衍生物2药物包合物的K 值。
[关键词] β2环糊精;包合物;包合常数;测定方法[中图分类号] R94 [文献标识码] A [文章编号] 1001-5094(2004)01-0019-06Methods of Determinating the Equilibrium Constants of Cyclodextrin or itsDerivative s ΠDrug Inclusion Complexe s and Their ApplicationW ANG Y a 2na 1, S UN Jun 2mei 2, Y U Li 2li 1, LU Y a 2peng 1, RE N Y ong13(11Center o f Drug Discovery ,Nanjing Normal Univer sity ,Nanjing 210097,China ;21School o f Medicine ,Hu 2bei Sanxia Univer sity ,Yichang 443001,China )[Abstract ] The methods of determinating the equilibrium constants of Cyclodextrin or its derivatives Πdrug in 2clusion com plexes and their application were summarized.Many kinds of equilibrium constants of inclusion com plexes of β2Cyclodextrin or its derivatives Πdrug reported in these years were introduced.[K ey w ords ] β2Cyclodextrin ,Inclusion com plexes ,Equilibrium constants of inclusion com plexes ,Method of determinating 环糊精具有“外亲水,内疏水”的特性,可以与药物及多种有机化合物形成包合物。
《环境仪器分析》第五章 紫外-可见吸收光谱法 (2)
碘钨灯:波长范围340-1200 nm。无论钨灯或碘钨灯, 在可见区发射的能量与工作电压4次方成正比,因此,预 使光源稳定,必须由一个很好的稳定电源。
紫外区:气体放电光源,如氢、氘灯。适用的波长 范围185~400 nm的连续光谱。
光栅是利用光的衍射与 干涉作用制成的,它可用 于紫外、可见及近红外光 域,而且在整个波长区具 有良好的、几乎均匀一致 的分辨能力。
优点:色散波长范围宽 、分辨本领高、成本低、 便于保存和易于制备等;
缺点:各级光谱会重叠 而产生干扰。
2019/10/31
6
3、样品室
样品室(吸收池,常用比色皿)
紫外区:必须是石英池 可见和近红外区:玻璃 池或石英池
2019/10/31
7
4、检测器(光电倍增管)
光
电子倍增极
敏
阴
极
电子倍 增极
光
R1
R2
R3
R4
负电压
阳
R
极
mA
R5
5、读数装置: 记录仪、数字显示器
2019/10/31
8
二、常用紫外-可见仪器类型
单光束紫外-可见分光光度计 双光束紫外-可见分光光度计 双波长分光光度计
例如:0.2M Na2SO4 溶解偶氮基—N=N—染料(甲基橙), 可以选择0.2 M Na2SO4作为溶剂参比。
2019/10/31
36
(2)试剂参比
如果显色剂或其他试剂在测定波长有吸收, 按显色反应条件下,只是不加入试样,同样加 入试剂和溶剂作为参比,可消除试剂中的组分 产生吸收的影响。
Fe2+ + 邻二氮菲 → 橙红色络合物
紫外_可见分光光度法在药物分析中的应用
第18卷第2期 分析科学学报2002年4月V ol.18 N o.2 JO U RN A L OF AN A L Y T ICA L SCI EN CE Apr. 2002文章编号:1006-6144(2002)02-0158-06紫外-可见分光光度法在药物分析中的应用郑 健 陈焕文 刘宏伟 李宝华 张寒琦 于爱民 金钦汉(长春分析仪器研究和技术开发中心,吉林大学化学系分析教研室,长春,130023)摘 要:本文着重评述了1998年以来国内紫外-可见分光光度法在药物分析中的应用,展望了其在该领域的发展趋势。
关键词:紫外-可见分光光度分析法;药物分析;综述中图分类号:O657.32;R917 文献标识码:A以分子吸收光谱为基础的紫外-可见区分光光度分析法具有设备简单、适用性广、准确度和精密度较好等特点,已在地质[1]、环境[2]、能源[3]、材料[4]、食品[5]等科学中发挥着重要作用,尤其是随着多元络合物、胶束增敏光度法、有机试剂等的发展,它已经成为应用最广泛的分析手段之一[6]。
分光光度法的早期应用集中在无机分析化学领域,即对为数众多的无机离子和化合物进行定性分析或定量测定[7]。
有机物的光度分析较无机物的开展晚,但发展十分迅速,已经从定性、半定量分析发展到定量分析,方法的灵敏度和选择性也有了很大的提高,能够用光度法进行分析测定的物质种类也在不断增多[8-10]。
近几年来,随着生命科学的发展,有机物光度分析的研究和应用热点主要集中在生物[11]、临床、药物[12]等方面。
光度法在药物分析中的应用历来受到大家的广泛关注和重视,世界各国都进行这一领域的相关研究[13,14]。
据统计,在药物分析中,分光光度法占29.1%,色谱法占25.5%,荧光、化学发光法占2.4%,与光度法有关的方法共计占31.5%,由此可见,光度法是药物分析最常用的方法之一[12]。
研究结果表明,光度法在药物分析中的可靠性可以和色谱法相蓖美[15],但其设备简单、操作方便、价格低廉、易于普及等特点是色谱法难于做到的。
日常紫外线强度 防护要求
日常紫外线强度防护要求紫外线指数大于2的时候需要防晒。
当紫外线指数在0-2之间时,紫外线强度最弱,通常在这个指数下是不会对肌肤造成伤害的,所以也就不需要给肌肤保护了。
但若是紫外线指数高于2,那么就需要根据具体情况选择合适的防晒方式来保护肌肤,防止肌肤被晒伤、晒黑。
万物的生长离不开太阳,当我们在夏日的海滩尽情嬉戏,在冬日的阳光下享受温暖的时候,不得不担心它会给我们带来烦恼。
特别是现在化学物质对于臭氧层的破坏,导致紫外线辐射增强,使皮肤癌的发生率增高,对人体健康造成极大影响,让我们对于享受阳光是“又爱又怕”。
首先我们要了解紫外线。
紫外线是太阳光中的一种,短时间照射会使皮肤晒红、晒黑,长时间照射则会导致皮肤出现黑斑、皱纹、老化,甚至引起皮肤癌。
夏季应每日关注天气预报中的紫外线指数,做好防护工作。
紫外线指数用数字表示:0~2:紫外线强度为一级,对人体影响较小,外出时可戴上太阳帽。
3~5:紫外线强度为二级:,外出时需要戴上太阳帽、太阳镜,涂防晒霜等。
5~6:紫外线强度为三级,外出时必须在阴凉处行走。
7~9:紫外线强度为四级:,在上午10点至下午4点这段时间内应尽量待在室内,避免外出。
≥10:紫外线强度为五级,应避免外出。
防晒的方法有哪些?1. 远离强紫外线。
每天上午10点到下午2点是一天当中紫外线最强的时段,应尽量避开这些时段外出。
2. 选择合适的防晒霜。
夏季的早、晚及阴雨天气可选用SPF(防晒指数)低于8的防晒霜。
中等强度阳光天气,防晒霜的SPF指数在8~15较好。
强烈阳光下,防晒霜的SPF指数应大于15。
3. 正确使用防晒霜。
应在出门前10~30分钟涂抹防晒霜。
脸部涂防晒霜的同时,切记不要忽略了下巴、脖子、耳朵、胳膊等部位。
在阳光强烈、长时间暴晒时,一般每2小时左右需要涂一次防晒霜,晚上卸妆后最好使用晒后护理品修复皮肤。
4. 合适的穿戴。
外出时最好穿浅色衣服,以棉麻为佳。
戴宽檐帽,尽量遮挡脸部、脖子等部位。
第二章 紫外-可见分光光度法-2
(3)温度的影响 在分光光度法测定中,通常都选用室温 显色反应。当温度对显色反应速度可能有较 大的影响时,需要考虑温度的影响。 合适的温度可用单因素实验来确定。
(4)显色时间 这里包括两种时间:一种是由于显色反 应速度不同,达到反应完全所需的时间;另 一种是有色化合物维持稳定的时间。 这两种时间均可用单因素实验来考察。
c. 快速扫描分光光度计陆续问世 利用光分析可以跟踪化学反应历程,一 般分光光度计只适于历程为20~30 min以上的 反应,要研究速度较快的反应,就需要设计 出快速扫描分光光度计,如:多道分光光度 计(采用:多道光子检测器,整个光谱扫描 时间不到1 s)。
4. 仪器的最新进展 (1) 仪器的自动化程度大大提高;
精确求取摩尔吸收系数的方法是:在 不同带通宽度时测定表观摩尔吸收系数, 绘制表观摩尔吸收系数对带通宽度的曲线 关系图,将曲线外推到带通宽度为零处, 这时相应的摩尔吸收系构造、类型及 发展趋势 1. 构造 通常由以下5个部分组成— (1) 一个或多个辐射源; (2)波长选择器; (3)试样容器 (吸收池) ; (4)辐射换能器; (5)信号处理器和读出装置。
对吸收池的要求:主要是能透过所研究的 光谱区辐射线。
吸收池的两个光学面必须平整光洁,使用 时不能用手触摸。
按材料可分为:玻璃吸收池和石英吸收池 两种。
吸收池有多种尺寸和不同结构,吸收池 的光径可在0.1 cm~10 cm之间变化,其中以 1 cm光径吸收池最为常用,根据使用要求 选用。 在用于高浓度或低浓度测定时,可相 应地采用光径较小或较大的吸收池。
(3) 蓝移 由于取代基或溶剂极性的影响,使吸收 谱带的最大吸收波长向短波方向移动的现象 称为短移、紫移或蓝移。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1)苯和取代苯 )
苯分子有3个共轭双键, 个成键轨道和3 苯分子有 个共轭双键,有3个成键轨道和 个共轭双键 个成键轨道和 个反键轨道, π→π*的跃迁情况比较复杂 的跃迁情况比较复杂。 个反键轨道, π→π 的跃迁情况比较复杂。 苯的吸收带: 苯的吸收带: E1带:184nm(ε约为 ×104),远紫外。 ε约为6× ,远紫外。 E2带:204nm (ε约为 ε约为7900),近紫外,有助色团 ,近紫外, 或生色团, 带红移,重要性大大增加。 或生色团,E2带红移,重要性大大增加。 B带:256nm (ε约为 带 ε约为200),气相或非极性溶剂中 , 呈现精结构,为芳香族化合物的重要特征。 呈现精结构,为芳香族化合物的重要特征。
2)稠环芳烃 )
稠环芳烃也有E 三个吸收带, 稠环芳烃也有 1,E2,B三个吸收带,同时 三个吸收带 具有伴随振转能级跃迁的精细结构。 具有伴随振转能级跃迁的精细结构。 稠环数增加,共轭体系增大,吸收带红移, 稠环数增加,共轭体系增大,吸收带红移, 吸收强度也增大。 吸收强度也增大。 三个吸收带红移幅度与稠环芳烃构型有关, 三个吸收带红移幅度与稠环芳烃构型有关, 线性排列的稠环三个吸收带红移的幅度不同, 线性排列的稠环三个吸收带红移的幅度不同,角 型稠环三个吸收带红移的幅度相似。 型稠环三个吸收带红移的幅度相似。
+
Cl
-
苯胺在酸性条件下 形成季胺盐,没有p 形成季胺盐,没有 电子与苯环共轭, 电子与苯环共轭, 紫外光谱几乎与苯 相同
生色团与苯环相连时, 带有较大的红移 带有较大的红移。 生色团与苯环相连时,B带有较大的红移。 出现强的K带 在200-250nm出现强的 带,ε>104,可能会 出现强的 带掩盖。 将B带,E2带掩盖。 带 苯及简单衍生物的紫外光谱特征, 苯及简单衍生物的紫外光谱特征,表2-8
273 268 268 λ max =非稠环二烯(a,b)+2 × 烷基取代+环外双键 =217+2×5+5=232(231)
λ max:232
2)α,β-不饱和羰基化合物 ) 不饱和羰基化合物
不饱和羰基化合物: α,β-不饱和羰基化合物:分子中含有与烯基 不饱和羰基化合物 共轭的羰基,包括α 不饱和酮、 共轭的羰基,包括α,β-不饱和酮、醛、酸、 不饱和酮 酯。 紫外光谱特征: 有一个强的K吸 紫外光谱特征:200-250nm有一个强的 吸 - 有一个强的 收带( 收带(εmax=1~2×104),由π→π 跃迁产生 × ),由π→π*跃迁产生 以上有一个n→π 产生的弱吸收R带 ;300nm以上有一个 →π 产生的弱吸收 带 以上有一个 →π*产生的弱吸收 小于100) (εmax小于 )
用该规则计算4个或 个一下的共轭烯烃的 用该规则计算 个或4个一下的共轭烯烃的 吸收带位置时 个或 个一下的共轭烯烃的K吸收带位置时 计算结果与实测值吻合。 ,计算结果与实测值吻合。 例1、计算 、 K带位置(λmax)。 带位置( 带位置 。
解:母体基本值:217nm 母体基本值: 取代烷基2个 取代烷基 个:2×5nm × λmax=227nm 实测值: 实测值:226nm
OH O
解:基本值 α位OH取代 个 取代1个 取代 计算值
215nm 35
β位烷基取代2个 2×12 位烷基取代 个 × 274 nm (实测值270nm) 实测值
1
例计算胆甾1,4-二烯酮 二烯酮 例计算胆甾 K吸收带的λmax 吸收带的λ 吸收带的
O
2 3 4
β’ β 5
β 该化合物的结构是一个交叉共轭,不需要对1,2位的双键和 该化合物的结构是一个交叉共轭,不需要对 位的双键和 基团进行校正。 β’ 基团进行校正。 解:基本值 2个β位烷基取代 个 1个环外双键 个环外双键 计算值 215nm 2× 12 × 5 244nm
超过4个以上双键的共轭多烯可以使用 超过 个以上双键的共轭多烯可以使用 Fieser-Kuhn规则。 规则。 规则 λmax = 114+15M+n(48.0-1.7n)-16.5Rendo10Rexo εmax=(1.74×104)n × N为共轭双键的数目,M为共轭体系上烷基 为共轭双键的数目, 为共轭体系上烷基 为共轭双键的数目 或类似烷基的取代基。 或类似烷基的取代基。Rendo为共轭体系中 为共轭体系中 带有桥环双键的环数目, 带有桥环双键的环数目,Rexo是环外双键的 是环外双键的 数目。 数目。
规则计算番茄红素的λ 例3,用Fieser-Kuhn规则计算番茄红素的λmax和εmax , 规则计算番茄红素的
个双键, 个共轭 个共轭, 个烷基取代, 解:化合物有13个双键,11个共轭,n=11;8个烷基取代, 化合物有 个双键 ; 个烷基取代 M=8;没有桥双键和环外双键。 ;没有桥双键和环外双键。 λmax=114+5×8+11×(48.0-1.7×11)-16.5×0× × × × 10×0=476nm (实测:474nm) 实测: × 实测 实测: εmax =(1.74×104)×11=19.1×104 (实测:19.1×104) × × × 实测 ×
3)芳香族杂环化合物 )
五员杂环、六员杂环、含杂原子的稠环。 五员杂环、六员杂环、含杂原子的稠环。 紫外光谱与苯系芳烃相似。 紫外光谱与苯系芳烃相似。 吸收带有精细结构。 吸收带有精细结构。 环上有助色团或生色团时吸收带红移。 环上有助色团或生色团时吸收带红移。
立体阻碍对紫外光谱的影响: 立体阻碍对紫外光谱的影响:
紫外光谱用溶剂 溶 剂 95% 乙 醇 水 正己烷 二 氯甲烷 四氢呋喃 四氯化碳 DMF 异丙醇 乙 腈 下限( ) 透 明 下限(nm) 210 210 210 235 220 265 270 210 210 溶 剂 乙醚 异辛烷 环己烷 二氧六环 氯 仿 苯 丙 酮 甲 醇 庚 烷 透 明下限 (nm) 210 210 210 230 245 280 330 215 210
OH
个双键, 个共轭 个共轭, 个烷基取代, 解:化合物有6个双键,6个共轭,n=6;6个烷基取代, 化合物有 个双键 ; 个烷基取代 M=6;2个环内双键,没有环外双键。 个环内双键, ; 个环内双键 没有环外双键。 λmax=114+5×6+6×(48.0-1.7×6)-16.5×2-10×0 × × × × × = 337.8nm εmax =(1.74×104)×6 = 10.44×104 × × ×
溶剂的影响
溶剂极性对异丙叉丙酮吸收带的影响
吸收带 正己烷 氯仿 K带 带 R带 带 230 329 238 315
甲醇 237 309
水 243 305
吸收带移 动规律
红移 蓝移
溶剂极性增强, β 不饱和羰基化合物的 带红移, 带蓝 不饱和羰基化合物的K带红移 溶剂极性增强,α,β-不饱和羰基化合物的 带红移,R带蓝 孤立羰基也有同样的溶剂效应。共轭烯烃的极性很小, 移。孤立羰基也有同样的溶剂效应。共轭烯烃的极性很小, 溶剂效应可以忽略。 溶剂效应可以忽略。
K带吸收的经验规律 带吸收的经验规律-Woodward规则 带吸收的经验规律 规则
基团 共轭双键的基本骨架 环内双烯 每增加一个共轭双键 每增加一个烷基或环烷基取代基 每增加一个环外双键 每一个助色团取代: 每一个助色团取代:RCOORORSCl或Br 或 R2N 贡献( ) 贡献(nm) 217 36 30 5 5 0 6 30 5 60
与助色团直接相连时,取代苯的 和 带红移 带红移, 与助色团直接相连时,取代苯的E2和B带红移,B 带的精细结构消失。 π共轭。 带的精细结构消失。p-π共轭。 苯酚在碱性条 H +
O O Na NaOH
件下电离为阴 离子, 离子,增加了 一个与苯环共 轭的孤对电子
H H N H NaOH
H N H
苯酰基化合物K带的位置计算法 苯酰基化合物 带的位置计算法
用表2-9的经验公式计算 甲氧基 用表 的经验公式计算6-甲氧基 的经验公式计算 蔡满酮的λmax 蔡满酮的λ
H3CO
O
解:基本值 邻位R取代 邻位 取代 对位OR取代 取代 对位 计算值
246nm 3 26 274nm (乙醇中实测值276nm) 乙醇中实测值
7
11
-8
2.3.4芳香族化合物 芳香族化合物
芳香族化合物含环状共轭体系,存在π→π*跃迁 芳香族化合物含环状共轭体系,存在π→π 跃迁,是 π→π 跃迁, 紫外光谱研究的重点
吸收带: 吸收带: R带: 含杂原子不饱和基团的 →π 跃迁所 →π* 带 含杂原子不饱和基团的n→π 产生,波长范围( 产生,波长范围(250-500nm),吸收强度比 , 较弱。 较弱。 K带:共轭双键的π→π 跃迁产生,波长范围 π→π* 带 共轭双键的π→π 跃迁产生, (210-250nm),吸收强度大。 ,吸收强度大。 B带:苯环的π→π 跃迁产生,波长范围( π→π* 带 苯环的π→π 跃迁产生,波长范围( 230-270nm),重心在 左右, ,重心在256nm左右,非极性 左右 溶剂中出现小峰或称精细结构, 溶剂中出现小峰或称精细结构,极性溶剂中 消失。 消失。
溶剂效应使α β 不饱和羰基化合物的吸收带位移 不饱和羰基化合物的吸收带位移, 溶剂效应使α,β-不饱和羰基化合物的吸收带位移, 使用表2-5的数值进行计算时需要做溶剂校正 使用表 的数值进行计算时需要做溶剂校正
溶剂校正表
溶剂 甲醇甲醇 乙醇 氯仿 二氧六 环 乙醚 正己烷 水
校正 值/nm
0
1
5
例2、计算该甾类化合物 带 、计算该甾类化合物K带 的最大吸收波长。 的最大吸收波长。
AcO
解:母体基本值:217nm 母体基本值: 增加2个共轭双键 × 增加 个共轭双键:2×30nm 个共轭双键 环内双键1个 环内双键 个:36nm 环内烷基取代5个 环内烷基取代 个:5×5nm × 环外双键3个 环外双键 个:3×5nm × RCO2-取代 个:0 取代1个 取代 λmax = 353nm 实测值: 实测值:353nm