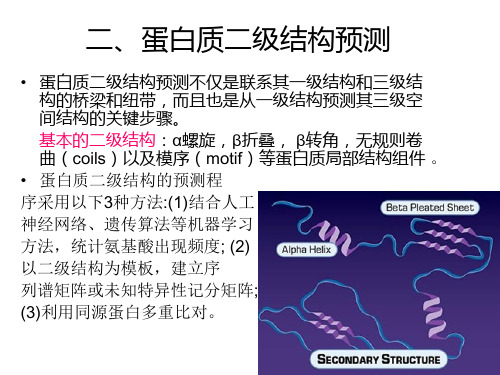
基因编码蛋白质的二级结构预测
蛋白质结构预测
序列基序识别 二硫键识别 折叠子识别 残基接触预测 结构域预测
结构表面识别
预测蛋白质表面结构功能关键区域
5
PredictProtein Secondary Structure
PredictProtein Secondary Structure
H:螺旋 E:折叠 L:环 e:暴露表面﹥16%残基 b:其它残基
3
PredictProtein提交界面
序列提交窗口
分析方法程序详解
PROFsec(默认) PROFacc(默认) 序列预测
基于轮廓(profile)的神经网络算法预测蛋 白质二级结构 基于轮廓(profile)的神经网络算法预测残 基溶剂可及性
PHDhtm(默认)
ASP(默认) COILS(默认) PROFtmb ProSite(默认) SEG(默认) PredictNLS(默认) DISULFIND(默认) AGAPE PROFcon ProDom(默认) CHOP ConSeq
22
SWISS-MODEL
• SWISS-MODEL是一个蛋白质3D结构数据库,库中收录的蛋白质结
构都是使用SWISS-MODEL同源建模方法得来的。
– /
• 基于同源建模法与PDB数据库已知结构的蛋白质序列比对 进行预测
23
SWISS-MODEL
蛋白质三维结构预测
方法 特点 工具
同源建模法 基于序列同源比对,对于序列相似度>30% SWISS-MODEL, CPHmodels ( Homology/Comparativ 的序列模拟比较有效,最常用的方法 e modelling ) 线串法/折叠识别法 (Threading/Fold recognition) 从头预测法 ( Ab initio/De novo methods ) “串”入已知的各种蛋白质折叠骨架内,适 于对蛋白质核心结构进行预测,计算量大 基于分子动力学,寻找能量最低的构象, 计算量大,只能做小分子预测
网格中基于结构分类和位矩阵编码并行遗传算法的蛋白质二级结构预测

Moen ) dl g 。蛋 白 质结 构 预 测 方 法 总 体 上 可 分 为 三 i
大 类 , 比 较 建 模 法 ( o p rt e moeig m t— 即 C m aai d l e v n h o ) 反 向折 叠 法 (n es lig 和从 头 预测 法 ( b d、 Iv r f dn ) eo a iiopeit nm t d 。 目前 , nt rdc o e o ) i i h 比较 建模 法 和反 向
ቤተ መጻሕፍቲ ባይዱ
能研 究 为核心 的“ 基 因组 时 代 ” 大 规 模 的结 构 基 后 ,
因组 、 白质组 以及 药物 基 因组 的研 究 计 划 已经 成 蛋
为新 的热 点 。每天 在 全 世 界 , 量 的 生物 学 数 据 不 大 断地 产生 和 积 累 , 有 效 地 处 理 、 存 和利 用 这 些 要 保 处 于异域 的 巨量信 息 , 须 建 立 强 大 而 高效 的信 息 必 处理 平 台 ,生 物信 息 网格” 运而 生 。 “ 应 蛋 白质结 构 预 测 是 生 物 信 息 学 中一 个 重要 问 题, 它研究 如 何 从 氨 基 酸 序 列 出 发 , 测 蛋 白质 的 预
结 构更 具 有非常 重要 的意 义 。 目前 对 蛋 白质 预测 的 大 部 分 研 究 主 要 是 基 于 对 已知 蛋 白质 结 构 规 律 的总 结 、 收 和利 用 , 用 吸 采 所 谓 的 、 于 知 识 的预 测 ( nw eg —ae rt n 基 K o l eb sd Poe d i
折 叠法 的研 究都 已取 得 了重 要 的进 展 , 两类 方 法 这 的共 同点是 : 预测 目标 蛋 白质 的结 构 时 都要 利 用 在
ncbi蛋白质序列的二级结构

ncbi蛋白质序列的二级结构
NCBI(National Center for Biotechnology Information)是一个国际知名的生物医学信息数据库,提供了大量的生物学、生物医学和基因组学等相关数据。
在NCBI数据库中,可以通过查询蛋白质的序列标识(如蛋白质的NCBI Accession号码)来获取该蛋白质的相关信息,包括二级结构信息。
获取蛋白质的二级结构信息可以通过以下步骤进行:
1. 在NCBI的主页(https:///)上的搜索栏中输入蛋白质的序列标识,点击搜索按钮进行搜索。
2. 在搜索结果页面中,找到与蛋白质相关的条目,点击进入对应的记录页面。
3. 在记录页面中,可以找到蛋白质的基本信息、序列信息等。
如果该蛋白质的二级结构信息可用,通常会在“Structure”或“3D structure”等部分提供相关链接。
4. 点击相关链接,可以进入蛋白质的二级结构数据库(如PDB,Protein Data Bank)或相关工具网站,以查看该蛋白质的二级结构信息。
需要注意的是,不是所有蛋白质的二级结构信息都可以在NCBI数
据库中直接获取,有些蛋白质可能没有经过结晶和测定结构的报道,或者相关信息尚未被整理和存储在数据库中。
此外,蛋白质的二级结构信息也可以通过其他生物信息学工具和数据库进行预测和推断。
蛋白质结构预测及其在结构生物学中的应用

蛋白质结构预测及其在结构生物学中的应用蛋白质是生命体内最基础的分子,是构成生命物质的基本单位。
每个蛋白质都具有一定的空间结构,这个结构决定了蛋白质的功能。
因此,蛋白质结构的预测对于生命科学、药物设计等领域具有重要的意义。
本文将探讨蛋白质结构预测的基本原理以及在结构生物学中的应用。
一、蛋白质结构预测的基本原理蛋白质的空间结构可以分为四个层次:一级结构(序列)、二级结构(α-螺旋、β-折叠等)、三级结构(多肽链的空间构象)和四级结构(多个多肽链之间的空间关系)。
预测蛋白质的空间结构,从根本上来说就是预测其三级结构的问题。
目前,大部分蛋白质结构预测方法都是基于基因组学的大规模测序数据的,结合一些现有的晶体结构数据库和序列数据库。
基本上,它们都大致包含以下三个步骤:第一步,根据蛋白质的氨基酸序列和模板数据,在蛋白质数据库中寻找最相似的结构;第二步,将找到的最相似的结构作为一个初始模型,使用蛋白质结构预测算法进行优化;第三步,选择最优解或者最优模型。
目前使用最广泛的预测方法是同源建模和蒙特卡罗模拟。
同源建模通过寻找蛋白质序列和已经被解析的蛋白质晶体或者核磁共振数据的相似性,构建一个已知的三维模型。
蒙特卡罗模拟是一种基于优化的预测方法,模拟蛋白质在空间中不同构象的状态,最后得到最佳的构象。
二、蛋白质结构预测在结构生物学中的应用蛋白质的结构预测对于结构生物学的发展起到了重要的推动作用。
此外,它还可以在多个领域中发挥重要的应用。
1.药物设计药物设计是利用化学或者生物学方法开发药品的过程。
在药物设计过程中,蛋白质结构预测是不可或缺的一步。
通过预测蛋白质的结构,科学家可以根据药物和靶标蛋白质之间的相互作用原理来精确设计和优化药物分子结构。
2.蛋白质工程蛋白质工程是指利用基因工程技术对蛋白质分子进行改造的过程。
结合蛋白质结构预测的结果,科学家可以从理论上探究如何改变蛋白质的某些特性,例如抗原性、稳定性、活性等,以实现特定的应用需求。
bioformatic website二级结构预测

卷曲螺旋分析
另一个能够直接从序列中预测的功能motif是α-螺旋的卷曲排列方式。在这种结构中,两种螺旋通过其疏水性界面相互缠在一起形成一个十分稳定的结构。
蛋白质卷曲的相关资源
资源 网址
coiled-coil /depts/biol/units/coils/coilcoil.htmlCOILS /software/COILS_form.htmlEpitopeInfo /Links.htm
4.几个序列保守结构域比较分析:/Tools/es/cgi-bin/clustalw2/ 输入格式:>名称,加 序列
/cl物理性质预测:
Compute PI/MW http://expaxy.hcuge.ch/ch2d/pi-tool.html
/interpro/scan.html
蛋白质的结构功能域分析
简单模块构架搜索工具(simple modular architecture research tool,SMART)一个较好的蛋白质结构功能域的数据,可用于蛋白质结构功能域的分析,所得到的结构域同时提供相关的资源的链接http://smart.embl-heidelberg.de/
最好的是/prosite)
蛋白质序列的(profile)分析
www.isrec.isb-sib.ch/software/PFSCAN_form.html
InterProScan综合分析网站
InterProScan是EBI 开发的一个集成了蛋白质结构域和功能位点的数据库,其中把SWISS-PROT,TrEMBL.PROTSITE.PRINTS.PFAM.ProDom等数据库提供的蛋白质序列中的各种局域模式,如结构域,motif等信息统一起来,提供了一个较为全央的分析工具。
基因预测的方法

基因预测的方法:(怎么样才能有一个感性认识?)方法1:最长ORF法将每条链按6个读码框全部翻译出来,然后找出所有可能的不间断开放阅读框(ORF),只要找出序列中最长的ORF,就能相当准确地预测出基因。
最长ORF法发现基因的一般过程(包括基因区域预测和基因功能预测2个层次):步骤1:获取DNA目标序列①如果已有目标序列,可直接进入步骤2;②可以通过PubMed查找感兴趣的资料,通过GenBank或EMBL等数据库查找目标序列。
步骤2:查找ORF并将目标序列翻译成蛋白质序列利用相应工具,如ORF Finder、Gene feature (Baylor College of Medicine)、GenLang (University of Pennsylvania)等查找ORF并将DNA序列翻译成蛋白质序列。
步骤3:在数据库中进行序列搜索利用BLAST进行ORF核苷酸序列和ORF翻译的蛋白质序列搜索。
步骤4:进行目标序列与搜索得到的相似序列的全局比对(global alignment)虽然步骤3已进行局部比对(local alignment)分析,但全局比对有助于进一步加深对目标序列的认识。
步骤5:查找基因家族进行多序列比对(multiple sequence alignment),获得比对区段的基因家族信息。
步骤6:查找目标序列中的特定模序分别在Prosite、BLOCK、Motif数据库中进行profile、模块(block)、模序(motif)检索。
步骤7:预测目标序列蛋白质结构利用PredictProtein(EMBL)、NNPREDICT(University of California)等预测目标序列的蛋白质二级结构。
步骤8:获取相关蛋白质的功能信息为了了解目标序列的功能,收集与目标序列和结构相似蛋白质的功能信息非常必要。
可利用PubMed进行搜索。
方法2:利用编码区与非编码区密码子选用频率的差异进行基因预测编码区的碱基组成不同于非编码区,这是由于蛋白质中20种氨基酸出现的概率、每种氨基酸的密码子兼并度和同一种氨基酸的兼并密码子使用频率不同(即密码子偏好)等原因造成的。
蛋白质结构与功能的预测方法总结和资料汇总

蛋白质结构与功能的预测方法总结和资料汇总“折叠(fold)”的概念“折叠(fold)”是近年来蛋白质研究中应用较广的一个概念,它是介与二级和三级结构之间的蛋白质结构层次,它描述的是二级结构元素的混合组合方式。
二级结构的预测方法介绍:Chou-Fasman算法:是单序列预测方法中的一种,它是使用氨基酸物理化学数据中派生出来的规律来预测二级结构。
首先统计出20种氨基酸出现在α螺旋、β折叠和无规则卷曲中出现频率的大小,然后计算出每一种氨基酸在这几种构象中的构象参数Px.构象参数值的大小反映了该种残基出现在某种构象中的倾向性的大小。
按照构象参数值的大小可以把氨基酸分为六个组:Ha (强螺旋形成者)、ha(螺旋形成者)、Ia(弱螺旋形成者)、ia(螺旋形成不敏感者)、ba(螺旋中断者)、Ba(强螺旋中断者)。
Chou和Fasman根据残基的倾向性因子提出二级结构预测的经验规则,要点是沿蛋白序列寻找二级结构的成核位点和终止位点。
这种方法可能能够正确反映蛋白质二级结构的形成过程,但预测成功率并不高,仅有50%左右GOR算法:也是单序列预测方法中的一种,因其作者Garnier, Osguthorpe和 Robson而得名。
这种方法是以信息论为基础的,也属于统计学方法的一种,GOR方法不仅考虑被预测位置本身氨基酸残基种类对该位置构象的影响,也考虑到相邻残基种类对该位置构象的影响。
这样使预测的成功率提高到 65% 左右。
GOR方法的优点是物理意义清楚明确,数学表达严格,而且很容易写出相应的计算机程序,但缺点是表达式复杂。
多序列列线预测:对序列进行多序列比对,并利用多序列比对的信息进行结构的预测。
调查者可找到和未知序列相似的序列家族,然后假设序列家族中的同源区有同样的二级结构,预测不是基于一个序列而是一组序列中的所有序列的一致序列。
基于神经网络的序列预测:利用神经网络的方法进行序列的预测,BP (Back-Propagation Network) 网络即反馈式神经网络算法是目前二级结构预测应用最广的神经网络算法,它通常是由三层相同的神经元构成的层状网络,使用反馈式学习规则,底层为输入层,中间为隐含层,顶层是输出层,信号在相邻各层间逐层传递,不相邻的各层间无联系,在学习过程中根据输入的一级结构和二级结构的关系的信息不断调整各单元之间的权重,最终目标是找到一种好的输入与输出的映象,并对未知二级结构的蛋白进行预测。
蛋白二级结构(卷曲螺旋)预测软件---------COILS的使用

预测结果图: 显示沿着序 列各个部分 形成卷曲螺 旋的倾向性
二、BCL-2基因在10个物种 Homo sapiens, Xenopus laevis, Gallus gallus ,Mus musculus, Bos taurus ,Rattus norvegicus ,Capra hircus ,Pan troglodytes ,Canis lupus familiaris ,Felis catus 中ClustalW序列比对分析、MEGA 系统进化分析 、MEME进行保守位 点分析。
BCL-2基因(即B细胞淋巴瘤/白血病-2 基因)是一种原癌基因,它具有抑制凋亡 的作用,并且近年来的一些研究已开始揭 示这一作用的机制。目前已经发现的BCL2蛋白家族按功能可分为两类,一类是像 BCL-2一样具有抑制凋亡作用,而另一类 具有促进凋亡作用。
BCL-2基因在10个物种中的位置:
>gi|262050657|ref|NM_001166486.1| Bos Taurus >gi|50950156|ref|NM_001002949.1| Canis lupus familiaris >gi|548519521|ref|XM_005697325.1| PREDICTED: Capra hircus >gi|57163882|ref|NM_001009340.1| Felis catus >gi|758371883|ref|NM_205339.2| Gallus gallus >gi|72198188|ref|NM_000633.2| Homo sapiens >gi|545477919|ref|NM_009741.4| Mus musculus >gi|694971627|ref|XM_001145537.2| PREDICTED: Pan testroglody >gi|8392973|ref|NM_016993.1| Rattus norvegicus >gi|225735550|ref|NM_001146093.1| Xenopus laevis
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
基因编码蛋白质的二级结构预测
在生命科学中,蛋白质是最基本的生物大分子之一,也是维持生命活动的基础。
蛋白质的功能和性质很大程度上取决于其三级结构,即由氨基酸组成的线性序列在生理条件下,经过特定过程形成的折叠构象。
然而,三级结构对绝大多数蛋白质的准确预测仍然具有很大挑战性。
相比之下,二级结构预测则更加简单并且通常可以得到很高的准确性。
什么是二级结构?
在蛋白质的氨基酸序列中,相邻的多个氨基酸之间通常存在一些规律的相互作用,从而形成了规则的空间结构。
其中最基本的结构单元是alpha螺旋和beta折叠。
这两种结构都具有稳定的形态和重复的配置,能够对蛋白质的稳定性和生物功能起到至关重要的作用。
如何预测二级结构?
预测蛋白质的二级结构是基于蛋白质中氨基酸的序列信息,通过建立序列与二
级结构之间的对应关系,预测氨基酸序列中会出现的螺旋和折叠段数量和位置。
目前主要采用的方法是通过机器学习算法挖掘氨基酸序列与已知二级结构之间的关联。
传统方法
传统方法主要包括一些机器学习算法,例如支持向量机(SVM)、神经网络(NN)、决策树(DT)等等。
这些算法的基本思想是采用特征向量表示氨基酸序
列的各种属性,然后通过训练与测试样本的比对,预测蛋白质二级结构。
这些算法在很大程度上可以提高二级结构预测准确度,但是其依赖于人工构建的特征向量,预测准确度在一定程度上受限。
深度学习方法
相比之下,深度学习方法在最近几年间得到了广泛的应用。
深度学习方法大大减少了特征向量的构建,从而可以获取更加本质的信息,从而提高了二级结构预测的准确度。
目前主要的深度学习方法包括卷积神经网络(CNN)、循环神经网络(RNN)和变换器(Transformer)等等。
这些算法可以对氨基酸序列进行端对端的处理,从而直接输出其二级结构预测结果。
与传统机器学习方法相比,深度学习方法在处理复杂问题方面更具优势,提高了二级结构预测的准确度。
结语
总之,蛋白质的二级结构预测是一项非常具有挑战性的研究课题,对于理解蛋白质三维结构和功能具有重要意义。
传统机器学习算法和深度学习算法都可以用于预测二级结构,不同方法各有利弊。
随着进一步科技发展,相信蛋白质结构预测算法的精度会不断提高,同时,也将对相关领域的应用产生重要影响。