分子模型的创建
化学实验教案:分子模型的构建

化学实验教案:分子模型的构建一、引言在化学教学中,分子模型的构建是一个非常重要的实验项目。
通过构建分子模型,我们可以直观地观察和理解分子的结构和性质,加深对化学概念的理解。
本实验教案将介绍如何利用简单的材料制作分子模型,并通过实践操作提高学生对分子结构的认识。
二、实验目的1. 了解并掌握分子模型的基本原理;2. 学会使用合适材料和方法制作分子模型;3. 提高学生对分子结构特征及其与化学反应之间关系的理解。
三、实验步骤1. 准备所需材料:彩色泡沫球(表示原子)、竹签(表示化学键)、卡纸等。
2. 将彩色泡沫球和竹签按照所需氢、氧原子等元素进行分类。
3. 挑选出需要构建的分子,在卡纸上绘制正确且比例合适的示意图。
4. 使用胶水将彩色泡沫球连接在一起,形成示意图上所示的分子结构。
5. 将竹签剪成合适长度,并将其插入泡沫球中,表示化学键。
6. 完成所有分子结构的制作后,让学生根据实际情况选择适当位置进行展示。
四、实验注意事项1. 实验时要小心操作,避免伤害到自己和他人。
2. 材料使用完毕后,要加以妥善保管,并及时清理工作区域。
3. 在选择材料和制作分子模型时,要尽量准确地符合实际分子结构的比例和形状。
4. 构建过程中,可以请学生根据需求调整泡沫球的大小或竹签的长度。
5. 将分子模型展示在明显可见的位置,可以增加学生对化学知识的认知。
五、预期结果通过本实验教案的指导和操作实践,在完成分子模型构建后,学生将能够更直观地理解分子结构、原子之间的化学键及其性质。
同时,通过展示分子模型,学生之间也能够互相分享知识与经验。
六、拓展应用1. 建议将本实验与其他相关的教学内容相结合,例如通过构建蛋白质或DNA 等大分子结构来拓展应用范围。
2. 鼓励学生主动利用已有知识和素材,尝试构建更复杂的分子结构,加深对化学概念的理解。
3. 提倡学生之间进行互动交流,分享自己独特的分子模型设计和构建经验。
七、实验结果分析通过本实验,学生在制作分子模型过程中可以直观地观察到原子之间的联系和结构。
分子模型晶体模型的制作
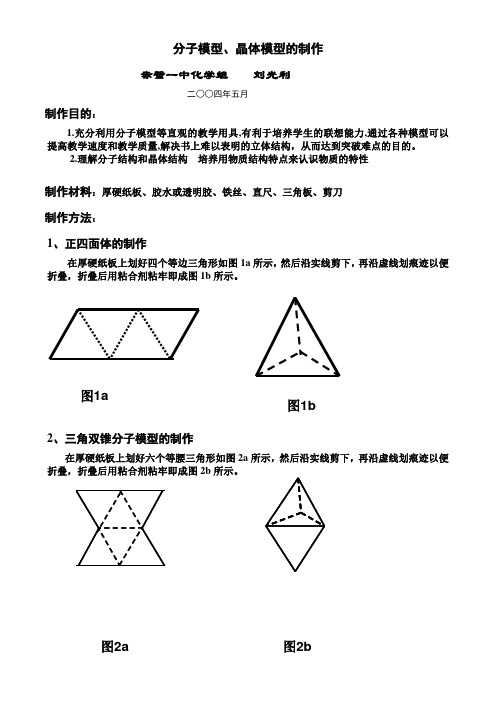
分子模型、晶体模型的制作赤壁一中化学组 刘光利二○○四年五月制作目的:1.充分利用分子模型等直观的教学用具,有利于培养学生的联想能力,通过各种模型可以提高教学速度和教学质量,解决书上难以表明的立体结构,从而达到突破难点的目的。
2.理解分子结构和晶体结构 培养用物质结构特点来认识物质的特性制作材料:厚硬纸板、胶水或透明胶、铁丝、直尺、三角板、剪刀制作方法:1、正四面体的制作在厚硬纸板上划好四个等边三角形如图1a 所示,然后沿实线剪下,再沿虚线划痕迹以便折叠,折叠后用粘合剂粘牢即成图1b 所示。
2、三角双锥分子模型的制作 在厚硬纸板上划好六个等腰三角形如图2a 所示,然后沿实线剪下,再沿虚线划痕迹以便折叠,折叠后用粘合剂粘牢即成图2b 所示。
图2a 图2b图1a图1b3、正八面体分子模型的制作在厚硬纸板上划好八个等边三角形如图3a所示,然后沿实线剪下,再沿虚线划痕迹以便折叠,折叠后用粘合剂粘牢即成图3b。
图3a 图3b4、正二十面体分子模型的制作(B12)在厚硬纸板上划好二十个等边三角形如图5a所示,然后沿实线剪下,再沿虚线划痕迹以便折叠,折叠后用粘合剂粘牢即成图5b所示。
图5a 图5b使用说明1.正四面体模型直接应用于白磷分子、甲烷分子、四氯化碳分子等正四面体分子结构的教学,也可应用于数学中立体几何的有关异面直线等方面的教学。
利用正四面体还可以组合成其他形状的立体图形。
例如,由一个正四面体可以切割成较小的正八面体,其方法是将正四面体的四个顶点从它的三条棱的中点切下,便可得到一个较小的正八面体。
如果以一个正四面体为中心,另用四个与之全等的正四面体分别与它的四个面相連接,就可以得到一个十二个面全等的凹十二面体。
2.三角双锥模型直接应用于五氯化磷(PCl5)等具有三角双锥结构的分子结构的教学。
也可用于数学教学。
3.正八面体应用于分子或离子组成为RX6、RX6n-型结构的教学。
两个或两个以上的正八机体之间还可以进行不同方式的重叠就可以得到多种空间图形,对讲解超八面体等空间结构教学有很大的帮助。
chemoffice20.0用法

chemoffice20.0用法ChemOffice20.0是一款强大的化学信息软件,它提供了全面的化学信息数据库,包括分子建模、反应机制、光谱分析等功能,广泛应用于化学、生物、材料等领域。
本文将详细介绍ChemOffice20.0的用法,帮助用户更好地掌握软件功能。
一、安装与启动ChemOffice20.0的安装过程较为简单,只需要按照安装向导的提示即可完成安装。
安装完成后,可以通过启动图标进入软件。
首次启动ChemOffice20.0时,会提示用户选择一个模板,以便进行分子建模和计算。
二、基本操作1.创建分子模型:在ChemOffice20.0中,可以通过点击“新建”按钮创建一个新的分子模型。
在创建过程中,可以选择不同的建模工具和参数,以满足不同的建模需求。
2.编辑分子模型:创建分子模型后,可以通过各种编辑工具对分子结构进行修改。
例如,可以使用“旋转”工具将分子模型旋转到正确的角度,以便观察分子的细节。
3.保存和导出分子模型:完成分子模型的编辑后,可以将其保存到本地或导出为常见的文件格式,如XYZ、MOL、PDB等。
三、化学信息查询ChemOffice20.0提供了丰富的化学信息查询功能,包括分子轨道、键能计算、光谱分析等。
用户可以通过软件内置的数据库查询所需的化学信息。
四、反应机制模拟ChemOffice20.0支持反应机制模拟,用户可以通过软件内置的反应模块构建反应路径,观察反应过程中的能量变化和产物生成。
五、光谱分析ChemOffice20.0提供了多种光谱分析工具,如红外光谱、核磁共振光谱、拉曼光谱等。
用户可以通过选择相应的光谱模块,导入分子模型,并设置相应的参数进行光谱分析。
六、软件优化与升级在使用ChemOffice20.0的过程中,可能会遇到一些性能问题或bug。
为了解决这些问题,用户可以参考软件官方论坛或社区,获取相应的优化方法或补丁程序。
同时,为了获得更好的使用体验和功能更新,用户可以定期检查软件的升级提示,及时进行软件更新。
化学实验教案:分子模型的构建

化学实验教案:分子模型的构建一、引言化学实验是帮助学生理解和掌握分子结构及其特性的重要手段之一。
在化学教学中,分子模型的构建被广泛应用于教学实践中,以帮助学生更好地理解分子结构与性质的关系。
本文将介绍如何编制一份高质量的化学实验教案,具体内容为分子模型的构建。
二、背景知识在进行分子模型的构建之前,首先需要向学生介绍有关原子和键的基础概念。
例如,原子是构成物质的最小单元,在化合物中通过共价键或离子键相互连接形成分子或离子晶格。
此外,对于常见元素的电子排布、共价键和离子键的推导也需逐步讲解。
三、实验目标实验目标是明确实验的目的并指导学生迈向预期结果。
我们可以设定以下几个实验目标:1. 了解原子间相互作用和空间排列对分子结构和性质的影响。
2. 学会使用简单材料搭建不同类型分子模型。
3. 探究不同类型分子模型之间存在的异同,并找出规律。
四、所需材料进行分子模型构建实验所需的材料一般可以在实验室或者学校化学教具库中找到。
以下是一些常见的所需材料:1. 带孔棍状球体或塑料模型球体2. 磁性原子模型(可使用磁铁吸附)3. 彩色软质小球(表示不同元素)4. 小磁铁(用于合成有机化合物)5. 硬纸板和剪刀(用于制作基座)五、实验步骤1. 介绍分子结构和键的概念,并让学生理解如何根据原子间的相互作用来搭建分子模型。
2. 根据提供的分子式,先由学生自主尝试搭建分子模型,然后与同组同学进行讨论,寻求最佳构建方法。
3. 学生们通过对比不同类型分子模型之间的异同以及其结构与性质之间的关系,推断出规律并展示给全班。
六、实验注意事项1. 实验过程中注意安全第一,避免导致任何伤害。
2. 细心处理小零件,避免损坏或误食。
3. 指导学生正确使用材料,并注意保存实验用具,以便下次实验使用。
七、效果评估在实施完分子模型构建实验之后,可以进行效果评估以了解学生对所学知识的掌握程度。
下面是一些建议的评估方式:1. 小组讨论:组织小组讨论,让学生分享他们对于分子模型构建过程中的发现和体会。
分子结构模型的构建及优化计算

分子结构模型的构建及优化计算分子结构模型的构建是化学研究和计算化学领域的重要一环,对于理解分子的性质和行为具有重要意义。
优化计算则是对构建的分子结构模型进行调整和优化,以求得最稳定和最符合实验结果的结构体系。
本文将介绍分子结构模型的构建方法以及常用的分子结构优化计算方法。
一、分子结构模型的构建1.实验室试验方法:实验室试验方法通过实验手段确定分子的构型和结构。
常用的实验方法包括谱学方法(如红外光谱、拉曼光谱、核磁共振等)、X射线方法和电子显微镜等。
这些实验方法可以提供分子的一些基本信息,例如键长、键角、晶胞参数等。
不过该方法需要实验设备和实验条件,有时也受到实验技术的限制。
2. 理论计算方法:理论计算方法主要通过量子力学计算、分子力学模拟和分子动力学模拟等,从基本粒子的角度计算分子的结构和性质。
在量子力学计算中,常用的方法有Hartree-Fock(HF)方法、密度泛函理论(DFT)方法、紧束缚模型(TB)方法等。
在分子力学模拟和分子动力学模拟中,常用的方法有分子力学(MM)方法、分子动力学(MD)方法等。
二、分子结构优化计算分子结构优化计算是对构建的分子结构模型进行调整和优化的过程,以找到最稳定和最符合实验结果的结构体系。
1.线性规划方法:线性规划方法是寻找一个解向量,使得目标函数最小或最大。
在分子结构优化计算中,可以通过线性规划方法来优化分子结构的内部参数,如键长、键角等。
2. Monte Carlo方法:Monte Carlo方法是一种通过随机抽样的方式来进行优化计算的方法。
在分子结构优化计算中,Monte Carlo方法可以通过随机调整分子的内部参数,以整个构象空间,寻找最稳定的构象。
3.遗传算法:遗传算法是通过模拟生物进化过程来进行优化计算的方法。
在分子结构优化计算中,可以将每一个分子结构看作一个个体,通过交叉、变异等操作模拟自然选择,以寻找最优解。
4.分子动力学模拟:分子动力学模拟是通过求解分子的运动方程,模拟分子的运动和变化过程。
中学化学教学中有效的分子模型建构方法

中学化学教学中有效的分子模型建构方法概述:化学是一门抽象而又具有实验性的科学,而分子模型则是化学中重要的概念之一。
分子模型的建构有助于学生理解化学现象和掌握化学知识。
本文将探讨中学化学教学中有效的分子模型建构方法,旨在提高学生的学习效果和兴趣。
一、球棍模型法球棍模型法是最常用的分子模型建构方法之一。
它通过使用不同颜色和大小的球代表原子,用棍子连接原子来表示化学键。
这种方法直观而简单,可以帮助学生理解分子的结构和化学键的形成。
例如,在讲解水分子的结构时,可以用两个红色球代表氧原子,用两个白色球代表氢原子,用棍子连接它们来表示水分子的构成。
这样的模型可以让学生更好地理解水分子的极性和氢键的形成。
二、立体模型法立体模型法是一种更为直观的分子模型建构方法。
它通过使用不同形状的物体来表示分子的结构,使学生能够更好地理解分子的三维形态。
例如,在讲解甲烷分子的结构时,可以使用四个等边三角形代表氢原子,一个正四面体代表碳原子,将它们组装在一起来表示甲烷分子的构成。
这样的模型可以让学生更加清晰地认识到分子的空间排布和键角的大小。
三、计算机模拟法随着科技的发展,计算机模拟法在化学教学中的应用越来越广泛。
通过使用化学模拟软件或在线分子模型构建工具,学生可以在电脑上进行分子模型的建构和观察。
这种方法不仅能够提供更多的分子结构选择,还能够模拟一些实验无法观察到的现象。
例如,在讲解有机物的立体异构时,可以利用计算机模拟软件构建不同的结构,并观察它们在空间中的排布和性质的差异。
这样的模拟实验可以让学生更加深入地理解分子结构与性质之间的关系。
四、实物模型法实物模型法是一种通过使用真实的物体来构建分子模型的方法。
这种方法可以让学生通过触摸和操作来更好地理解分子的结构和性质。
例如,在讲解离子化合物的结构时,可以使用磁性球和棒子来表示阳离子和阴离子,将它们组装在一起来构建离子晶体的结构。
这样的实物模型可以让学生更加直观地感受到离子间的吸引力和排列规律。
分子模型的制作

图 4a
B12
图4b
正四面体与正八面体之间的演变
C60(足球烯)
凹十二面体
正十二面体
足球烯示意图
足球烯展开图型
足球烯展开图形
足球烯示意图
分子模型的制作方法
赤壁一中化学组 刘光利
制作目的:
1.充分利用分子模型等直观的教学用具,有利于培养学生 的联想能力,通过各种模型可以提高教学速度和教学质量,解 决书上难以表明的立体结构,达到突破重、难点的目的。 2.理解分子结构和晶体结构 培养用物质结构特点来认识 物质的特性
制作材料:
厚硬纸板、胶水或透明胶、铁丝、直尺、三角 板、剪刀
图2a
图2b
3.正八面体分子模型的制作(XY6型)
• 在厚硬纸板上划好八个等边三角形如图3a 所示,然后沿实线剪下,再沿虚线划痕迹 以便折叠,折叠后用粘合剂粘型的制作(B12模型)
• 在厚硬纸板上划好二十个等边三 角形如图4a所示,然后沿实线剪 下,再沿虚线划痕迹以便折叠, 折叠后用粘合剂粘牢即成图4b所 示。
1.正四面体分子模型的制作(白磷)
• 在厚硬纸板上划好四个等边三角形如图1a所示, 然后沿实线剪下,再沿虚线划痕迹以便折叠, 折叠后用粘合剂粘牢即成图1b所示。
图1a
图1b
2.三角双锥分子模型的制作(PCl5型)
• 在厚硬纸板上划好六个等边三角形如图2a所示,然 后沿实线剪下,再沿虚线划痕迹以便折叠,折叠后 用粘合剂粘牢即成图2b所示。
武汉大学分子模拟实验作业第七章分子结构模型创建和优化计算
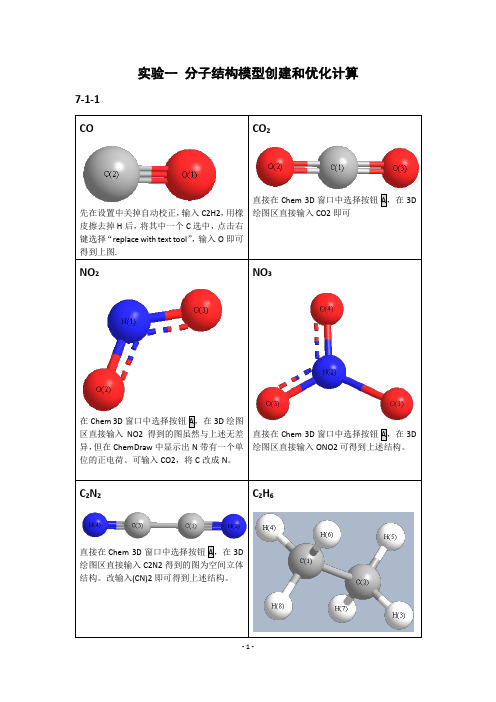
实验一分子结构模型创建和优化计算7-1-1绘图区直接输入C2H6即可。
HCOOH直接在Chem 3D窗口中选择按钮A,在3D绘图区直接输入HCOOH即可。
3DH3PO47-2-1-7-3-1 MM2O 和O 距离为2.765AE 1=-5.4331Kcal/Mol E 2= 0.0289 Kcal/Mol D=E 1-2E 2=-5.3753 Kcal/MolHF/6-31++GO 和O 距离为3.0 ÅE1= -95408.4 Kcal/mol (-152.04313 Hartrees) E2== -47701.8 Kcal/mol (-76.01774 Hartrees) D=E1-2E2=-4.8 Kcal/mol = (-0.00765 Hartrees) MP2/6-31++GO 和O 距离为2.9 ÅE1=-95650.8 Kcal/mol (-152.42932 Hartrees) E2=-47822.3 Kcal/mol (-76.20978 Hartrees) D=E1-2E2=-6.2 Kcal/mol (-0.00976 Hartrees) DFT= B3LYPO 和O 距离为2.8 ÅE1= -95870.8 Kcal/mol (-152.77997 Hartrees) E2= -47932.5 Kcal/mol (-76.38545 Hartrees) D=E1-2E2=-5.8 Kcal/mol (-0.00907 Hartrees)MOPAC-PM3O 和O 距离为3.0 ÅE1= -108.7 Kcal/molE2=-53.4 Kcal/molD=E1-2E2=-1.9 Kcal/mol7-4-2”化学意义”:RuClClPNN简介:第二代Grubbs催化剂是Grubbs在1999年对第一代催化剂的改进。
Grubbs 通过系统地对催化剂结构-性能关系进行研究,发现催化剂的活性与其中一个膦配体的解离有关,认为催化循环过程中经过一个高活性的单膦中间体,然后才与烯烃发生氧化加成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Massage with Ghost amtos
Ghost atoms may also be used for a counterpoise calculation for an estimate of the magnitude of BSSE, counterpoise corrections provide only a crude estimate and not an upper bound on the error). A counterpoise correction can be achieved by specifying the dimer structure with the atomic symbol for one monomer replaced by a ghost atom. Since ghost atoms have no basis functions by default, they must be explicitly added via the ExtraBasis facility or a general basis set. See also the discussion of Massage in the manual.
# HF/6-31G* Massage Test
HF + H2O interaction energy: HF removed 01 X H 1 1.0 F 2 rHF 1 90.0 O 2 rHO 1 90.0 3 180.0 H 4 rOH 2 aHOH 1 90.0 H 4 rOH 2 aHOH 5 180.0 rHF 0.9203 rHO 1.8086 rOH 0.94 aHOH 126.4442 1 Nuc 0.0 2 Nuc 0.0
内坐标和直角坐标的转换
Molden 读、存 Chem3D Gaussian View
Guassian 计算结果输出
在Link 0 Commands 行加入: MOND Hyperchem MERCURY WLViewerPro4.2 Molview MOLEKEL4.3 *** Amber Babel
end
1.400000 1.089000 120.000
虚原子(Dummy Atoms)
Dummy atoms are pure mathematic points, and are useful in defining geometries 表示做:X,xx 等。 程序将其原子序数为 -1 鬼原子(Ghost Atoms) A ghost atom can be useful to specify the location of the off-nucleus basis functions, which have zero nuclear charge and mass. 表示做:Bq,G,Gh 等。
内坐标(Internal Coordinate or Z-matrix)
用键长、键角、二面角等几何参 量来表示分子中原子间的键长和位置 的一种坐标方式。矩阵中的每一行对 应于分子内一个原子的内坐标。
文件格式(syntax):
原子标号,原子1,键长,原子2,键角,原子3,二面角
Label,Atom 1,Bond-length,Atom2,Bond-angle,Atom3,Dihedral-angle (Torsion angle)
构建分子模型
数据形式:
直角坐标(Cartesian Coordinate) 内坐标(Internal Coordinate or Z-matrix)
创建数据方法:
X-ray衍射晶体数据 手工建立 利用工具软件, 如Chem3D
数据形式:
直角坐标(Cartesian Coordinate)
1 cc2 1 hc3 1 hc3 2 hc3 2 hc3 2 hcc3 2 hcc3 1 hcc3 1 hcc3
3 180.000 3 0.000 3 -180.00
cc2 hc3 hcc3
1.400000 1.089000 120.000
Gamess z-matrix
zmat angstroms c c 1 cc2 h 1 hc3 2 hcc3 h 1 hc3 2 hcc3 h 2 hc3 1 hcc3 h 2 hc3 1 hcc3 variables cc2 1.400000 hc3 1.089000 hcc3 120.000 constants end
程序将其原子序数为 0
USING GHOST ATOMS
Ghost atoms provide a convenient way to request arbitrary points at which to compute electrostatic properties. These points can be specified directly in Cartesian coordinates in the standard orientation. It is sometimes easier to specify the points in internal coordinates, and, since properties are automatically computed at all nuclear coordinates, ghost atoms can be added to the Z-matrix at points of interest.
Atomic charges with hydrogens summed into heavy atoms: 1 1 C -0.143082 2 H 0.000000 3 H 0.000000 4 C 0.000000 5 H 0.000000 6 H 0.000000 7 Bq 0.143082 Sum of Mulliken charges= 0.00000
#P B3LYP/6-31G* GFINPUT IOP(6/7=3) TEST Example of NBO bond orders
0 1 C 0.000000 H 0.919278 H -0.919239 C 0.000000 H -0.919278 H 0.919239 Gh 0.700000
3,1, 3,2, 3,1, 3,2, 3,1, 3,2,
ADF Z-matrix
atoms z-matrix c c1 h1 h1 h2 h2 cc2 2 hc3 2 3 hc3 1 3 hc3 1 3 hc3
hcc3 hcc3 hcc3 hcc3
180.000 0.000 -180.00
cc2 hc3 hcc3
常见内坐标文件: Gaussian (.gjf)
Gamess(.inp)
ADF
Mopac(.mop .dat) Note:
0o < Bond-angle < 180 o
-180o ≤ Dihedral-angle ≤180 o
Gaussian Z-matrix
c c h h h h
0.665676 1.237739 1.237787 -0.665676 -1.237739 -1.237787 0.700000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
Total atomic charges: 1 1 C -0.286170 2 H 0.143082 3 H 0.143087 4 C -0.286170 5 H 0.143082 6 H 0.143087 7 Bq 0.000000 Sum of Mulliken charges= 0.00000
文件格式(syntax):
Label, x, y, z [例]乙烯的直角坐标文件格式(*.xyz)
6 C 0.000000 C 0.000000 H 0.943102 H 0.943102 H -0.943102 H -0.943102 0.000000 0.000000 0.000000 1.335000 0.000000 -0.544500 0.000000 1.879500 0.000000 1.879500 0.000000 -0.544500
3 180.000 3 0.000 3 -180.00
Mopac Z-matrix
SYMMETRY
C C H H H H 0.000000 0 1.400000 1 1.089000 1 1.089000 0 1.089000 0 1.089000 0 4 4 5 5 6 6 0.000000 0 0.000000 0 0 0 0 0.000000 0 0.000000 0 1 0 0 120.000000 1 0.000000 0 1 2 0 120.000000 0 180.000000 0 1 2 3 120.000000 0 0.000000 0 2 1 3 120.000000 0 -180.000000 0 2 1 3
Gaussian GAMESS-US GAMESS-UK
MOPAC, AMPAC
Interfaced via Molden Format
ADF MOLPRO ACESII MOLCAS JAGUAR DALTON HONDO CADPAC
Note: / 量子化学软件中文网
MOLDEN: a pre- and post- processing program for molecular and electronic structures
Interfaced via program output
Massage
The Massage keyword requests that the molecule specification and basis set data be modified after it is generated. The standard basis functions are assigned to atoms before Massage alterations take place, while the number of electrons is computed from the atomic numbers after the modifications.