第三章 序列两两比对
实验3 两条序列比对与多序列比对
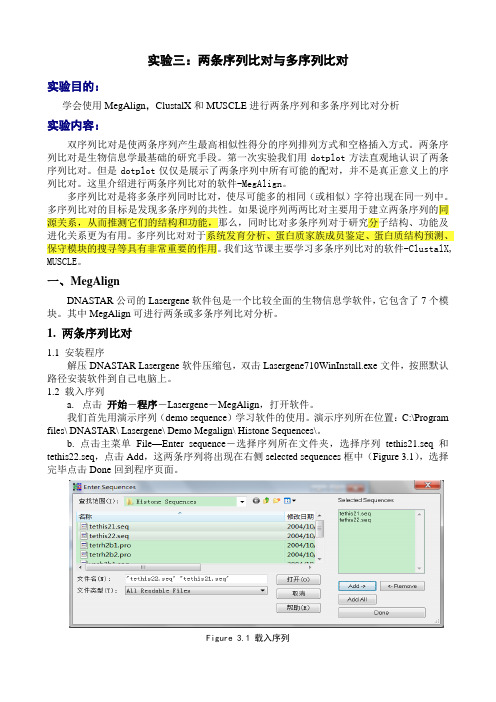
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
第三章 序列相似性比较

序列比对问题
基因在进化中存在插入/缺失突变,序列比对时应该 将这些考虑这些突变,以便获得到更好的对齐结果。
ATGCATGCATGCATGCATATATATATATATATATGCATGCATGCATGCATGC | | | | | | | | | | | | | || | | | | | | | | | | || | | | | | | | | | | | CGATCGATCGATCGATATATATATATGCATATATATGCATGCATGCATGCAT
等价矩阵 BLAST矩阵 转移矩阵 7 31 6
t= ACACACTGA Alignment-2 s= ACACAC-CA |||||| | t= ACACACTGA
7 31 2
氨基酸计分矩阵
氨基酸计分矩阵 —— 等价矩阵 —— 遗传密码矩阵 —— 疏水矩阵 —— PAM矩阵 —— BLOSUM矩阵
ATGCATGCATGCATGCATATATATATATATATATGCATGCATGCATGCATGC | | | || | | | | | | | | | | | CGATCGATCGATCGATATATATATATGCATATATATGCATGCATGCATGCAT
ATGCATGCATGCATGCATATATATATATATATATGCATGCATGCATGCATGC | | | | | | | | | | | | | || | | | | | | | | | | || | | | | | | | | | | | CGATCGATCGATCGATATATATATATGCATATATATGCATGCATGCATGCAT
CGATCGATCGATCGATATATATATATGCATATATATGCATGCATGCATGCAT
3-2,生物信息学序列比对

当序列差异较大时,上述问题更加明显。
例如
三条序列:
Seq1: ARKCV Seq2: ARCV Seq3: AKCV
若Seq1,2先比对, 再加入Seq3:
ARKCV AR-CV A-KCV ARKCV A-RCV A-KCV
ARKCV AR-CV AK-CV
Seq1,3先比对,再 加入Seq2: Seq2,3先比对,再 加入Seq1:
rij 2 lg 2(q / e)
BLOSUM62打分矩阵
BLOSUM & PAM
序列相似性与PAM及BLOSUM矩阵的大致 对应关系:
序列相似性 % PAM数值
99 1 90 11 80 23 70 38 60 56 50 80 40 30 20
112 159 246
BLOSUM数值
90
渐进比对
每条序列的权值
ClustalW的打分原则
Score:BLOSUM62的分数
ClustalX的使用
1. FASTA序列格式,多序列:
ClustalX的使用 ——导入序列文件
执行比对
文件导出
多序列比对:结果处理
BioEdit, GeneDoc等软件
GeneDoc软件,导入.aln文件
PAM矩阵
71个蛋白质家族的1572种变化; 序列相似性 > 85%; 功能同源的蛋白质 通过中性进化,引入 可接受的点突变; 进化模型:
A. 基本假设:中性进化,Kimura,1968; B. 进化的对称性: A->B = B->A; C. 扩展性:通过对较短时间内氨基酸替代关系 的计算来计算较长时间的氨基酸替代关系;
BLOSUM62矩阵构建步骤:
序列对齐分析ppt课件

点,从而探索导致它们产生共同功能的序列模式 3、蛋白质序列与核酸序列相比 探索核酸序列可能的表达框架;把蛋白质序列与具有
三维结构信息的蛋白质相比,从而获得蛋白质折叠 类型的信息。 4、查询序列与整个数据库的所有序列进行比对 从数据库中获得与其最相似序列的已有的数据
PDB序列,除了EST、STS、 GSS和
0,1,2阶段的HTGS序列
month nr中过去30天的最新序列
dbest 非冗余的Genbank+EMBL+DDBJ+ PDB的EST部分
dbsts 非冗余的Genbank+EMBL+DDBJ+ PDB的STS部分
htgs 未完成的0,1,2阶段的高产量基因组序列 (3阶段完成的HTG序列在nr库里)
pdb
PDB结构数据库中的蛋白质序列
yeast 酵母基因组中编码的全部蛋白质
E.coli 大肠杆菌基因组中编码的全部蛋白质
Kabat Kabat的免疫学相关蛋白质序列
alu
由REPBASE中的Alu重复序列翻译而来,用来遮
蔽查询序列中的重复片段
7
BLAST的核酸数据库
数据库 简 述
nr
非冗余的GenBank+EMBL+DDBJ+
10
NCBI的BLAST网址是: /BLAST/
11
第二节 多序列比对
一、概念 多序列比对:把两条以上可能有系统进化关系的
序列进行比对的方法。 进行多序列比对后可以对比对结果进行进一步处
理,例如构建序列模式的profile,将序列聚类 构建分子进化树等等。 二、软件 目前使用最广泛的多序列比对程序是 CLUSTALW(它的PC版本是CLUSTALX)。
东北农业大学生命科学学院生物信息学课件第三章第二节序列两两比对算法

第三章序列比较第二节序列两两比对算法1. 序列两两比对基本算法直接方法—生成两个序列所有可能的比对,分别计算得分(或代价)函数,然后挑选一个得分最高(或代价最小)的比对作代价)函数然后挑选个为最终结果。
本质问题:优化动态规划算法(Dynamic Programming)依据动态规划寻优策略→Needleman-Wunsch算法Needleman Wunsch最短路径问题C1起点终点W1C2W2C1+1路径1:C1 + w1 ?路径2:C2 + w2 ?取最小值!算法求解:从起点到终点逐层计算利用动态规划方法求解序列的两两比对起点终点A ATTC………CGAAG AGTC GAAGGT AGTC………GAAGG ATTC………CGAAG A 1AGTC………GAAGG +T()AATTC CGAAG AATTC………CGAAG AGTC………GAAGG +-T (2)ATTC………CGAAG +A (3)TAGTC………GAAGG-求解过程AATTC………CGAAG起点终点TAGTC………GAAGG•从两个序列前端开始•逐步推进•直到两个序列的末端。
中间过程:比对:S:i与0:T:j序列S: i-1 i i+1序列t: j-1 j j+1序列S: i-1 i i+1 Case1:匹配(si ,tj)序列t: j-1 j j+1序列S: i-1 i i+1序列t: j-1 j j+1序列S: i-1 i i+1Case2:序列t: j-1 j j+1删除(s i ,-)序列S: i-1 i i+1序列t: j-1 j j+1序列S: i-1 i i+1Case3:序列t: j-1 j j+1插入(-,t j )s t m n 。
设序列、的长度分别为和考虑两个前缀0:s:i0:t:jt 所有较短子序列的最优比对即已知假如已知序列0:s:i 和0:t:j 所有较短子序列的最优比对,即已知:(()()1)0:s:(i-1)和0:t:(j-1)的最优比对(2)0:s:(i-1)和0:t:j 的最优比对(3):s:和:t:(j-1)的最优比对0i 0(j 1)则0:s:i 和0:t:j 的最优比对一定是上述三种情况之一的扩展((这取决于(1)替换(s i ,t j )或匹配(s i ,t j ),这取决于s i 是否等于t j ;(2)删除(s i ,-);(3)插入(-,t j )。
第三章序列对比与数据库搜索(上)

滑动窗口技术与完整点矩阵图结果比较
(a) (1)对人类(Homo sapiens) 与黑猩猩(Pongo pygmaeus) 的β球蛋白基因序列进行比较的 完整点阵图。
(1)
(2) (b) (2)利用滑动窗口对以上的 两种球蛋白基因序列进行比较 的点阵图,其中窗口大小为10 个核苷酸,相似度阈值为8。
两条序列的相似程度计算
相似度: 它是两个序列的函数,其值越大,表示两个序列越相似 。 距离: 两个序列之间的距离越大,则两个序列的相似度就越小。
序列转化与字符编辑操作(Edit Operation)
• 字符编辑操作可将一个序列转化为一个新序 列 匹配 Match(a,a) 删除 Delete(a,-) 替代 Replace(a,b) 插入 Insert(-,b)
符号 G A 含义 G A 说 明 Guanine Adenine 鸟嘌呤 腺嘌呤
T
C R Y
T
C G or A T or C
Thymine
Cytosine Purine Pyrimidine
胸腺嘧啶
胞嘧啶 嘌呤 嘧啶
M
K S W H B V
A or C
G or T G or C A or T A or C or T G or T or C G or C or A
Alignment1: GACGGATTAG GATCGGAATAG
Alignment2: GA CGGATTAG GATCGGAATAG
序列表示与字母表
• 字母表
– 4字符DNA字母表:{A, C, G, T} – 扩展的遗传学字母表或IUPAC编码 – 单字母氨基酸编码
扩展的遗传学字母表或IUPAC编码
3序列比对原理
100个残基发生一次替换的Dayhoff’s PAM-1矩阵
针对不同的进化距离采用PAM 矩阵
序列相似度 = 40% | 打分矩阵 = PAM120
50% | PAM80
60% | PAM 60
PAM250
→ 14% - 27%
第三节 序列比对算法 Dotplot算法
评估两条序列相似度最简单的方法之一是利用点阵图。
• 假设两条序列:CACGA和CGA,使用统一的空位和 失配罚分 • 则:1、给第一条序列加一个空位
2、给第二条序列加一个空位 3、两条序列都不加空位
如果知道了ACGA与GA最佳比对的得分,就可以立即计算出表中第一行的 得分。同样地,如果知道了表中第二、第三行剩余序列的最佳比对的得 分,就可以计算出起始位点的不同的三种比对得分。
(a)Leabharlann (b)(a)对人类(Homo sapiens)与黑猩猩(Pongo pygmaeus)的β球蛋 白基因序列进行比较的完整点阵图。(b)利用滑动窗口对以上的两种球 蛋白基因序列进行比较的点阵图,其中窗口大小为10个核苷酸,相似度阈 值为8。
常用对比软件:BLAST(bl2seq)
动态规划: Needleman 和 Wunsch 算法
第三章 序列 比对原理
Principles of Sequence Alignment
• Biology
- What is the biological question or problem?
• Data
-What is the input data? -What other supportive data can be used ?
• 考虑这样的两条核苷酸序列: AATCTATA和AAGATA 仅有三种比对方式
生物信息学 第三章:序列比对原理
blastx Search protein database using a translated nucleotide query tblastn Search translated nucleotide database using a protein query
tblastx
Search translated nucleotide database using a translated nucleotide query
一、序列比对打分
序列分析的目的是揭示核苷酸或氨基酸序列编码的 高级结构或功能信息目的。
二、打分矩阵
稀疏矩阵、相似性打分矩阵
(一)DNA打分矩阵
即两个序列中相应的核苷酸相同,计1分;否则计0分。 如果考虑颠换和置换,可采用以下打分矩阵
(二) 氨基酸序列打分矩阵
1.PAM矩阵(Dayhoff突变数据矩阵)
(二)直系同源、旁系同源
直系同源基因(orthologous gene)是指在不同物种 中有相同功能的同源基因,它是在物种形成过程中 形成的。
旁系同源基因(paralogous gene)是指一个物种内 的同源基因。
直系同源基因和旁系同源基因统称为同源基因 (homolog)。
第二节 序列比对打分方法
ClustalX的比对步骤: 1.加载要比对的序列文件 序列格式可以支持NBRF/PIR、EMBL/Swiss-Ppot、 FASTA、GDE、Clustal、MSF、RSF等。
2.多序列比对
3.比对结果输出
(二)T-Coffee工具 (三)MultAlin工具 (四)MAFFT工具
比对结果
(二)高级BLAST工具 1、PSI-BLAST(position specific interated BLAST)
实验3 两条序列比对与多序列比对
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
(完整)生物信息学复习小结(中科大)
第二章:序列的采集和存储2. 序列数据的存储核酸序列数据库国际三大核酸序列数据库:GenBank, EBML, DDBJdbEST: Expressed Sequences Tags数据库UniGene等RefSeq: The Reference Sequence Database蛋白质序列数据库UniProtSwiss—prot & TrEMBL, PIR基因组数据库: Ensembl第三章序列比对I序列间比对的对应关系:匹配、替代、缺失、插入双序列比对算法:Dot matrix(点阵法)动态规划算法Needleman-Wunsch算法Sij = max of Si—1,j-1 + σ(xi , yj )Si—1,j —d ( 从左到右)Si,j—1 —d ( 从上到下)Smith-Waterman 算法Sij = max of 0Si-1,j-1 + σ(xi , yj )Si—1,j -d (从左到右)Si,j—1 -d (从上到下)FASTA和BLAST算法PSI-BLAST (位点特异性迭代BLAST):1. 使用普通的blast算法进行搜索;2。
将搜索得到的序列,包括输入的序列放在一起,构建位点特异性的矩阵(Position Specific Matrix);3。
利用上面得到的矩阵谱(profile),再次在数据库中进行搜索;4. 重复2 ,3 步,直到不再有新的序列出现;PHI—BLAST : 模式发现迭代BLAST第三章序列比对Ⅱ打分矩阵及其含义1,计分方法2, PAM系列矩阵3, BLOSUM 系列矩阵多序列比对:方法改进1。
渐进方法:代表:ClustalW/X, T—Coffee(1)ClustalW/X:计算过程1。
将所有序列两两比对,计算距离矩阵;2. 构建邻接进化树(neighbor—joining tree)/指导树(guide tree);3。
将距离最近的两条序列用动态规划的算法进行比对;4。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
4
序
言
识别序列的进化关系能帮助我们描绘未知序列的功能。当一组序列 的比对显示出了显著的相似性,我们就认为它们属于同一个家族。如果 这个家族中一个成员的结构或功能已知,那么它的这些信息就可以推广 到家族中其它没有通过实验验证的序列。所以序列比对可以用于预测未 知结构和功能的序列的结构和功能。 序列比对可以用来推断两条序列是否是相关的。如果两条序列显著 相似,那么这种相似性是随机产生的可能性非常小,也就是说这两条序 列有共同的进化起源。当一个序列比对被正确的做出来,它就反应了两 条序列的进化关系:相同位置出现不同残基的区域代表残基替换;一条 序列的残基对应另一条序列的空位的区域代表在进化的过程中一条序列 出现过残基插入或删除。有一种情况也是可能的,那就是来源于同一祖 先的两条序列在某种程度上分离以至于它们有共同祖先的关系已经不能 从序列的水平上识别了。如果那样的话,它们的进化距离就必须通过其 它的方法识别了。
7
序
言
序列相似(similarity)与序列一致(identity)
序列比对中用到的另一对相关术语是序列相似与序列一致。这两个 概念对于核苷酸序列是同义的。而对于蛋白质序列,这两个概念是非常 不同的。在蛋白质序列比对中,序列一致是指待比对的两条序列中相同 残基匹配的比例;序列相似是指待比对的两条序列中很容易彼此替换具 有相似理化性质残基匹配的比例。有两种方法计算序列相似/一致度。 一种方法是用两条序列的全部长度,而另一种是利用较短的序列进 行标准化。第一种方法用如下公式计算序列相似度: S=[(Ls*2)/(La+Lb)]*100 其中S是序列相似的百分比,Ls是相似的残基数目,La和Lb分别是两条 序列的长度。
8
序
言
序列相似(similarity)与序列一致(identity)
第一种方法用如下公式计算序列一致度: I=[(Li*2)/(La+Lb)]*100 其中I是序列一致的百分比,Li是一致的残基数目,La和Lb分别是两条序 列的长度。 第二种方法利用如下公式计算序列的一致/相似度: I(S)%=Li(S)/La% 其中La是较短序列的长度。
Dotmatcher(bioweb.pasteur.fr/seqanal/interfaces/dotmatcher.html) Dottup(bioweb.pasteur.fr/seqanal/interfaces/dottup.html) Dothelix(www.genebee.msu.su/services/dhm/advanced.html) MatrixPlot(www.cbs.dtu.dk/service/MatrixPlot/ )
18
序列比对的方法
自我比较
19
序列比对的方法
点阵方法
点阵法给出了两条序列关系的一种直观描述,它很容易识别出序 列中高度相似的区域。这种方法的一个显著的优点是可以基于矩阵中 存在的水平方向上或垂直方向上长度相同的对角线来识别序列中的重 复区域。因此这种方法在基因组学中有一定应用。它在识别染色体重 复和比较两个高度相关的基因组中基因顺序的保守性非常有用。它在 通过检测序列的自补性来识别核苷酸序列的二级结构也非常有用。 点阵法能显示所有可能的序列匹配。然而,它要求用户将邻近的 对角线连接起来来构造带有残基插入和删除的全序列比对。这种方法 的另一个缺点是它缺少评价比对质量的统计上的精确性。这种方法在 两两比对中也存在局限性。它很难构造多序列比对。下面是几个利用 点阵法设计的序列两两比对的工具。
20
序列比对的方法
动态规划方法
动态规划是一种通过匹配两条序列中所有可能的字符对来确定最 优比对的方法。它和点阵法基本相似,就是也需要构造一个二维的矩 阵。而它确是通过将点阵转换为记录序列间匹配和失配的得分矩阵来 找最优比对的定量的方法。通过寻找矩阵中的最高分数集合来精确的 找到最佳联配
21
序列比对的方法
6
序
言
序列同源(homology)与序列相似(similarity)
对于DNA序列,由于只存在四种碱基,两条不相关的序列同一位置 至少有25%的机会相同,而对于蛋白质序列,一共有20中氨基酸,所以 不相关的序列中出现同一氨基酸的概率为5%。序列长度也是一个关键因 素。序列越短随机出现相同的残基的概率就高,序列越长随机相同的概 率就越小。 这就要求对短的序列给予高的阈值来判断它们同源。例如,对于有 100个氨基酸的蛋白质序列,如果全局比对有30%或更高的残基相同,就 认为它们很有可能同源,这个范围被称为“安全范围”;如果有20%~ 30%的氨基酸相同,它们的同源关系就不那么肯定了,这个范围被称为 “模糊范围”;如果有低于20%的氨基酸相同,那么就很难认为它们具 有同源关系,这个范围被称为“黑暗范围”(P33图3.1)。这是一种不精 确的方法,尤其是对模糊范围很难判定是否同源,后面介绍的序列比对 的统计学上的显著性将会给出精确的方法判断序列是否同源。
22
序列比对的方法
A
A T
1 0 0 0 0
C
0 1 1 2 2
T
0 2 2 2 3
G
0 2 3 3 3
C
0 2 3 4 4
C
0 2 3 5 5
T
0 3 3 5 6
G
C T
最佳比对是:A C T G C C T
A -TG - CT
23
序列比对的方法
空位罚分
在寻找最优比对时要用到代表插入和删除的空位。因为在自然进化 过程中插入和删除发生的频率要比替换相对少,所以引进空位在计算上 应该是比较谨慎的以反映在进化中插入和删除发生的少。然而实际中分 配罚分值是比较任意的,因为没有革命性的理论来确定引进插入和删除 所付出的精确的代价。如果罚分值设置的过低,空位就会过多以至于无 关序列也会得到很高的相似性得分;如果罚分值设置的过高,空位就很 难被引进以至于很难找到合理的比对,这也是不切实际的。通过对球状 蛋白的经验学习,我们已经得到了一组适合于大部分比对的罚分值。在 大多数比对程序中它们可以被用作默认的罚分值。
12
序列比对的方法
13
序列比对的方法
14
序列比对的方法
点阵方法
用点阵法比较大序列时存在一个问题,那就是高噪音水平。在大多 数点平面上,点被画的到处都是,使真正的比对难以被识别。对于DNA 序列,问题尤其严重,因为DNA序列中只有四种字符,所以每一个碱基 都有1/4的机会与另一条序列的碱基匹配。为了降低噪音,我们不再直接 扫描单个碱基的匹配,而是引进过滤技术。用一个适当长度的“窗口” 来覆盖一段连续的残基。当使用过滤器时,窗口沿着两条序列滑动来比 较所有可能的连续残基。只有当等于窗口长度的一段连续残基和另一条 序列相同长度的残基匹配时才在矩阵中相应位置画点。这种方法在降低 噪音水平上是有效的。窗口也被称作“tuple”,其大小是可以被操纵的以 使它能识别特定模式的序列。然而,如果窗口被选的很长,比对的敏感 性就降低了。
序列比对的方法
比对算法
比对算法,不管是全局比对还是局部比对,基本上是相似的,只是 比对相似残基时最优化策略不同。所有比对算法都是基于以下三种方法 的:点阵方法,动态规划方法和基于单词的方法。点阵法和动态规划算 法在这章讲,而基于单词的方法将在下一章讲。
11
序列比对的方法
点阵方法
最基本的序列比对方法是点阵法,也叫点平面图法。这是一种在二 维矩阵中比较两条序列的直观方法。待比较的两条序列被放在矩阵的横 轴和纵轴上。我们通过扫描一条序列上的每一个残基与另一条序列的所 有残基的相似性来比较两条序列。如果发现了一个残基匹配就在相应位 置画一个点。而矩阵的其它位置保持空白。如果两条序列有大量的相似 区域,就用直线沿着对角线将连续的点连接起来。如果对角线的中间出 现断点,就表明有残基的插入和删除。矩阵中平行的对角线代表序列中 的重复区域。
3
序
进化基础
言
DNA和蛋白质是进化的产物。它们可以被认为是编码数百万年 进化史的分子化石。在进化史上,这些分子经历了随机变化过程,期 中一些被进化所选择而保留了下来。这些被选择的序列逐渐积累突变 和交叉,进化的痕迹在序列的某些部分被保留下来从而可以识别它们 共同的祖先。进化痕迹的存在是由于一些对序列结构和功能起关键作 用的残基倾向于被自然选择所保留;而另一些不起关键作用的残基倾 向于频繁的改变。例如,一个酵母家族的活性位点残基倾向于被保存 下来是由于它们对催化功能起作用。所以,通过序列比对,保守的和 改变了的序列模式就能被识别出来。在比对中序列的保守度体现了不 同序列之间的进化关系。反之,序列之间的差别反映了在进化的过程 中序列以替换、插入和删除残基的形式发生了变化。
15
序列比对的方法
16
序列比对的方法
17
序列比对的方法
点阵方法
点阵法有许多变形。例如,一条序列可以和它自身比对以识别内部 重复元素。在自比对当中会存在一条主对角线以表示其完美匹配。如果 内部重复元素存在,会观察到在主对角线的上方或下方有短的对角线。 DNA序列的自补(也叫反向重复),例如那些存在发夹结构的家族,也 能用点距阵法识别。在这种情况下,一条DNA序列与它的反向补序列进 行比较。平行的对角线代表反向重复。为了比较蛋白质序列,必须使用 一个权重系统来描述氨基酸残基的相似度。
9
序列比对的方法
序列两两比对的最终目的是找到两条序列的最佳匹配,也就是找到 残基之间的最大相似。为了达到这个目标,一条序列需要相对于另一条 序列移动以找到具有最大相似程度的位置。有两种经常用到的不同的比 对策略:全局比对和局部比对。
全局比对和局部比对
在全局比对中,我们假定两条序列在整个长度上是相似的。全局比 对时我们从头到尾的比较两条序列以找到最佳匹配。这种方法很适用于 比对两条大体上长度相同且极度相似的序列。对于发散的不同长度的序 列,这种方法不能产生最理想的结果因为它不能识别出两条序列中高度 相似的局部序列。 在局部比对中,我们不假设两条序列全局相似,只是找两条序列中 高度相似的局部区域而不考虑其它区域。这种方法能比对比较分散的序 列来找出DNA或蛋白质序列中的保守模式。被比对的两条序列可以不等 长。 这种方法很适合于比对包含相似模块的分散的生物序列, 以找出 domain或motif。 10