酶切位点的保护碱基
酶切位点保护碱基

0
GGAATTC CATATG GAATTCC
75
>90
GGGAATTC CATATG GAATTCCC
75
>90
Nhe I
GGCTAGC C
0
0
CG GCTAGC CG
10
25
CTA GCTAGC TAG
10
50
—
Not I
TT GCGGCCGC AA
0
0
ATTT GCGGCCGC TTTA
10
10
AAATAT GCGGCCGC TATAAA
10
10
ATAAGAAT GCGGCCGC TAAACTAT
25
90
AAGGAAAAAA GCGGCCGC AAAAGGAAAA
25
>90
1r
Nsi I
TGC ATGCAT GCA
10
>90
CCA ATGCAT TGGTTCTGCAGTT
>90
>90
f
>90
>90
TT GGCGCGCC AA
>90
>90
1
Ava I
CCCCGGG G
50
>90
CCCCCGGG GG
>90
>90
TCC CCCGGG GGA
>90
>90
1
BamH I
CGGATCC G
10
25
CG GGATCC CG
>90
>90
CGC GGATCC GCG
>90
>90
'Bgl II
酶切位点保护碱基

0
0
0
25
0
50
75
>90
Pst I
GCTGCAGC
0
0
TGCACTGCAGTGCA
10
10
AACTGCAGAACCAATGCATTGG
>90
>90
AAAACTGCAGCCAATGCATTGGAA
>90
>90
CTGCAGAACCAATGCATTGGATGCAT
0
0
Pvu I
CCGATCGG ATCGATCGAT TCGCGATCGCGA
10
>90
10
>90
0
50
0
50
Sph I
GGCATGCC CATGCATGCATG ACATGCATGCATGT
0
0
0
25
10
50
Stu I
AAGGCCTT GAAGGCCTTC AAAAGGCCTTTT
>90
>90
>90
>90
>90
>90
Xba I
CTCTAGAG GCTCTAGAGC TGCTCTAGAGCA CTAGTCTAGACTAG
>90 >90 >90
>90 >90 >90
25 >90 >90
0 >90 >90
0 0 >90
10
0 50 >90
0 0 >90 50
>90 >90 >90
>90 >90 >90
Hind III
酶切位点所加保护碱基
BamHI
1
97 (2)
LITMUS 29
HindIII
BglII
3
100 (2)
LITMUS 29
NsiI
BsiWI
2
100 (2)
LITMUS 29
BssHII
BspEI
2
1
100 (1)
8 (2)
LITMUS 39
LITMUS 38
BsrGI
BsrGI
BsrGI
2
1
99 (2)
88 (2)
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。取1 µg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。反应缓冲液含70 mMTris-HCl (pH 7.6),10 mMMgCl2,5 mMDTT及适量的NaCl或KCl(视酶的具体要求而定)。20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
>90
50
0
0
>90
50
EcoR I
GGAATTCC
CGGAATTCCG
CCGGAATTCCGG
8
10
12
>90
>90
>90
>90
>90
>90
Hae III
GGGGCCCC
AGCGGCCGCT
TTGCGGCCGCAA
8
10
12
>90
>90
>90
>90
>90
>90
Hind III
各种酶切位点的保护碱基引物设计必看

各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
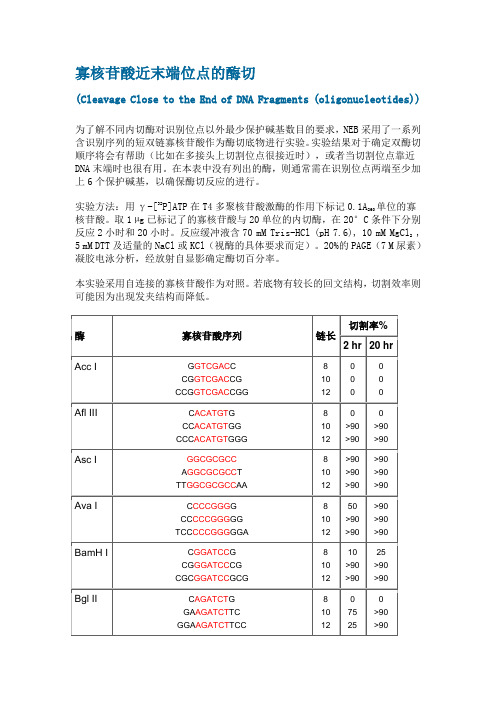
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH , 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer 完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
各种酶切位点的保护碱基 引物设计必看
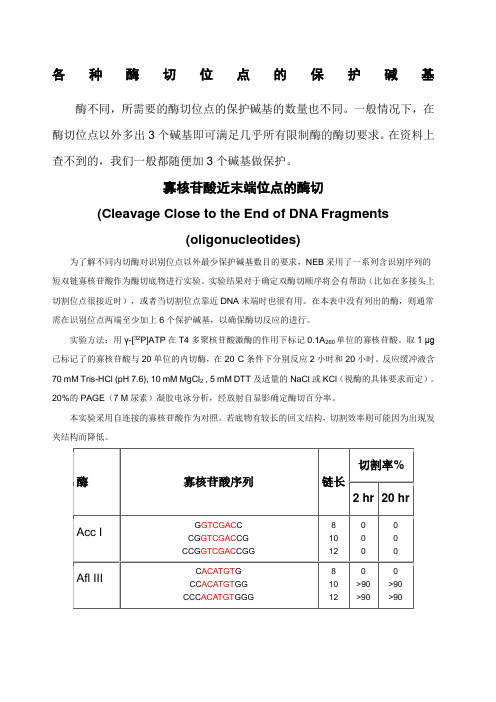
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
PCR设计引物时酶切位点的保护碱基

PCR设计引物时酶切位点的保护碱基引物设计是PCR实验的关键步骤之一,引物的好坏会直接影响到PCR反应的成功与否。
而在引物设计过程中,酶切位点的保护碱基是需要考虑的重要因素之一在PCR实验中,引物的作用是指定PCR反应的放大区域,并提供启动位点供聚合酶结合。
一般情况下,引物至少需要包含一段特定的DNA序列,以便与目标序列互补配对。
在引物设计过程中,选择合适的酶切位点是十分必要的。
酶切位点是指位于特定DNA序列上的限制酶可以识别并切割的区域。
酶切位点的选择通常需要考虑如下几个方面:1.切割效果:选择切割效果好的酶切位点可以提高PCR反应的特异性和灵敏度。
经典的选择是选择一种具有4-6个碱基的酶切位点,并且该位点在引物中间的位置。
这可以有效防止酶切位点的保护碱基对PCR反应的影响。
2.特异性:引物需要选择适合的酶切位点,以确保只有目标序列被放大,而不包括其他与之相关的非特异性序列。
因此,在选择酶切位点时应尽量避免与其他非特异性序列存在相似性。
3.引物长度:引物长度的选择也与酶切位点相关。
如果引物长度过短,可能会导致酶切位点过于靠近PCR反应产物的端点,从而使切割效果不佳。
因此,在引物设计时,应选择适当的引物长度,以保证酶切位点的保护碱基不会对PCR反应产物的生成产生不利影响。
酶切位点的保护碱基是指在特定的DNA序列上,通过选择相应的碱基来避免受到酶切的影响。
常见的保护碱基有甲基化碱基、磷酸化碱基以及接上阻断扩增的非互补碱基等。
1.甲基化碱基:将酶切位点中的一些碱基进行甲基化处理,可以有效地阻止特定酶的切割作用。
甲基化碱基可以通过DNA甲基转移酶进行甲基化修饰。
2.磷酸化碱基:磷酸化碱基是在引物设计过程中添加磷酸基团的方法,通过给酶切位点添加一个磷酸基团来阻断酶的切割作用。
3.非互补碱基:为了阻断酶切位点的切割作用,可以在酶切位点的周围引入一个与其不互补的碱基序列。
这样可以阻断酶的结合和切割。
总的来说,选择合适的酶切位点和保护碱基对PCR实验的成功至关重要。
各种酶切位点的保护碱基引物设计必看
各种酶切位点的保护碱基引物设计必看Document serial number【KK89K-LLS98YT-SS8CB-SSUT-SST108】各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH , 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
各种酶切位点的保护碱基引物设计必看
各种酶切位点的保护碱基引物设计必看酶切位点是指特定的序列,酶可以识别并在该位置切割DNA分子。
这些位点的特异性使得酶在分子生物学中广泛应用于DNA片段的定位和切割。
然而,在一些实验中,我们可能需要保护酶切位点周围的碱基,以免酶切,并且只在特定的位置引导酶切。
因此,保护碱基引物的设计对于实验的成功非常重要。
以下是保护碱基引物设计的一些建议。
首先,保护碱基引物的设计需要考虑引物的长度。
引物的长度通常为18到30个碱基,具体的长度需要根据实验的需求和酶切位点周围的序列特征来确定。
引物的长度应足够长,以确保引物和靶序列的特异性,但不应过长,以免引物形成二级结构或与非特异性位点结合。
其次,保护碱基引物的设计需要考虑引物的碱基组成。
在设计引物时,建议尽量避免引物中出现酶切位点周围的碱基序列,以防止酶的误切。
例如,如果我们希望保护酶切位点周围的AATTC序列,可以设计一个引物,其中没有AATTC序列。
同时,引物的碱基组成应尽量避免多聚核苷酸或含有GC碱基的片段,以防止引物之间的结合或引物与非特异靶序列的结合。
此外,保护碱基引物的设计需要考虑引物的特异性。
在设计引物时,建议使用特异性的引物序列,以确保引物只与目标酶切位点结合。
可以通过使用生物信息学工具,如BLAST,来验证引物的特异性。
引物的特异性还可以通过调整引物的长度和碱基组成来进一步提高。
最后,保护碱基引物的设计需要考虑引物的热力学性质。
引物的热力学性质包括引物的熔解温度(Tm值)和引物之间的配对。
引物的Tm值与引物的碱基组成、长度和引物与靶序列之间的碱基配对相关。
可以使用在线工具,如NEB的Tm计算器,来计算引物的Tm值,并对不同的引物进行比较。
此外,引物之间的配对可以通过设计引物的末端序列来调整,例如末端的碱基配对或非配对等。
总结起来,保护碱基引物的设计需要考虑引物的长度、碱基组成、特异性和热力学性质。
通过合理设计引物,可以保护酶切位点周围的碱基,并在特定位置引导酶切,为实验的成功提供有力的保障。
酶切位点保护碱基
酶切位点保护碱基-PCR引物设计用于限制性内切酶酶切反应来源:easylabs 发布时间:2009-11-08 查看次数:12704本文给出了分子克隆中常用限制性内切酶的保护碱基序列,如AccI,A flIII,AscI,AvaI,BamHI,BglII,BssHII,BstEII,BstXI,ClaI,E coRI,HaeIII,HindIII,KpnI,MluI,NcoI,NdeI,NheI,NotI,N siI,PacI,PmeI,PstI,PvuI,SacI,SacII,SalI,ScaI,SmaI,S peI,SphI,StuI,XbaI,XhoI,XmaI,为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A2 60单位的寡核苷酸。
各种酶切位点的保护碱基
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 µg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer 完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。