Ectopic over-expression of two apple Flowering Locus T homologues,
The expression levels of the translational factors eEF1A 12 correlate

Research paperThe expression levels of the translational factors eEF1A 1/2correlate with cell growth but not apoptosis in hepatocellular carcinomacell lines with different differentiation gradeG.Grassi a ,1,B.Scaggiante b ,*,1,R.Farra a ,B.Dapas a ,F.Agostini a ,D.Baiz b ,N.Rosso c ,C.Tiribelli b ,caDepartment of Clinical,Morphological and Technological Sciences,Division of Internal Medicine,University of Trieste,Via Giorgieri,1,34127Trieste,ItalybDepartment of Biochemistry,Biophysics and Macromolecular Chemistry,University of Trieste,Via Giorgieri,1,34127Trieste,ItalycCentro Studi Fegato,AREA Science Park,Campus Basovizza,34012Trieste,ItalyReceived 5March 2007;accepted 10July 2007Available online 20July 2007AbstractDespite the involvement of the elongation factors eEF1A (eEF1A1and eEF1A2)in the development of different cancers no information is available on their possible contribution to the biology of hepatocellular carcinoma (HCC).We investigated the expression of both forms of eEF1A in HepG2and JHH6cell lines considered to be a good in vitro model of HCC at different stage of differentiation.Our data indicate that the mRNA amount of eEF1A1is increased in both cell lines as compared to normal liver tissue,but eEF1A2mRNA level is markedly increased only in JHH6.Moreover,the less differentiated cell line JHH6displays higher EEF1A1and EEF1A2mRNAs levels and an higher nuclear-enriched/cytoplasm ratio of EEF1A protein compared to the better differentiated HepG2cell line.Over-expression depends only partially on gene amplification.The more abundant mRNA levels and the higher nuclear-enriched/cytoplasm ratio of eEF1A in JHH6neither correlate with apoptosis resistance nor with proliferation rate in sub-confluent cells.However,in confluent cells,a clear tendency to maintain JHH6into the cell cycle was observed.In conclusion,we document the increased mRNA levels of EEF1A genes in HCC cell lines compared to normal liver.Additionally,we show the increased nuclear-enriched/cytoplasmic protein ratio of eEF1A and the marked raise of the expression of both eEF1A forms in JHH6compared to HepG2,suggesting the possibility that eEF1A forms might become a relevant markers related to HCC tumor phenotype.Ó2007Published by Elsevier Masson SAS.Keywords:Hepatic cancer cells EEF1A2;EEF1A1;Apoptosis;Cell cycle1.IntroductionThe eukaryotic elongation factor 1A (eEF1A)is a protein which has been long recognized to play a key role in pro-tein translation [1].It carries aminoacyl-tRNA as complexeEF1A-GTP-aa-tRNA to the A site of the ribosome,following correct codon-anticodon recognition and hydrolysis of GTP.Several EEF1A genes are present in the human genome,but the eEF1A protein is encoded only by two actively transcribed genes:EEF1A1and EEF1A2[2].EEF1A1maps on chromo-some 6q14.1,whereas EEF1A2maps on chromosome 20q13.3and the two coding sequences share 78%of homol-ogy with 92%of protein identity.In mammalians,eEF1A1protein is ubiquitously expressed with the exception of skeletal muscle,heart and brain,where during terminal differentiation eEF1A1declines and eEF1A2protein is produced [3].*Corresponding author at:Molecular Biology Section,Department of Bio-chemistry,Biophysics,and Macromolecular Chemistry,University of Trieste,Via Giorgieri,1,34127Trieste,Italy.Tel.:þ390405583678;fax:þ390405583691.E-mail address:scaggiante@bbcm.units.it (B.Scaggiante).1These two authors contributed equally to this paper.0300-9084/$-see front matter Ó2007Published by Elsevier Masson SAS.doi:10.1016/j.biochi.2007.07.007Available online at Biochimie 89(2007)1544e1552/locate/biochiBeside the central role in protein translation,eEF1A is implicated in several biochemical processes including the interaction with the cytoskeleton[4e6],apoptosis and cell proliferation.With regard to apoptosis,it has been shown that eEF1A2ectopic over-expression protects muscle cells from caspase-3induced apoptosis[7],and its absence in mice results in an increased muscle cell apoptosis[8]. eEF1A1protects also against apoptosis induced by IL-3 withdrawal[9],and the acquisition of cisplatinum resistance in human head and neck cancer cell line is associated to eEF1A1over-expression[10].eEF1A interconnection with cell proliferation derives from the observations that eEF1A levels directly correlates to cell cycling rate[11e13].Addi-tionally,redistribution from the cytoplasm to the nucleus of eEF1A was observed in proliferating compared to starved cells [14].The eEF1A ability to modulate apoptosis and cell growth has strictly linked this protein with cancer.Recently,eEF1A has been implicated in the process of oncogenic transforma-tion.The tumorogenic potential of eEF1A1has been proposed following the observation that its over-expression correlates with increased metastatic potential in mammary adenocarci-noma[15].The ectopic expression of eEF1A2induces the transformation towards a malignant phenotype of mouse and ratfibroblast[16]and its expression in a human ovarian cell line that does not express eEF1A2confers a more aggressive neoplastic phenotype compared to the parental cell line[17]. Finally,amplification of the genome region encoding for eEF1A2protein has been found in breast,colon and ovary tumors[18e20].Whereas the oncogenic role of eEF1A has been proposed in different tumors where its over-expression has been docu-mented[10,21,22],no information is so far available on its role in the biology of hepatocellular carcinoma(HCC).To address this issue,HepG2and JHH6cell lines,derived by HCC but displaying a different phenotype[23,24],were examined in terms of expression levels of eEF1A1/2,gene amplification and relation to apoptosis and cell growth.2.Materials and methods2.1.Cell culturesHepG2and Hela cell lines were cultured in Dulbecco’s modified Eagle’s medium(DMEM)(Euroclone,Celbio, Devon,UK);JHH6and HuH7[23],obtained from Japan Health Science Research Resources Bank(HSRRB, JCRB1030depositor Dr.Nagamori,S),were cultured in William’s medium E(Sigma-aldrich,St Louis,MO)and Dulbecco’s modified Eagle’s high glucose medium DMEM (Euroclone,Celbio,Devon,UK).All media contained10% fetal bovine serum(FBS),2mM L-glutamine,100U/ml penicillin and100m g/ml streptomycin(Euroclone,Celbio, Devon,UK).HepG2,HuH7and JHH6were assigned to high,medium and low hepatocytic differentiation grade on the base of the capacity to synthesize albumin,a known marker of hepatic differentiation[25].HepG2and HuH7displayed3.6Æ1.6 and1Æ0.1albumin mRNA content,respectively,whereas no albumin mRNA was detected in JHH6(data not shown). HuH7cell lines derives from a differentiated hepatoma whereas undifferentiated morphology was observed in JHH6 [23].Despite the almost undetectable albumin production, JHH6were able to synthesize ferritin,thus showing a residual hepatic phenotype(data sheet).2.2.RNA and DNA extractionTotal RNA and DNA were extracted from2Â106cells cultured near confluence or from two normal liver tissues obtained from male donors of58and68years old during surgical resection for angioma by using Tri-reagent procedure (Sigma Chem.Co.St.Louis,MO)and suspended in50m l of DEPC water or in an adequate volume of8mM NaOH, respectively.The quality and integrity of total RNA and DNA were evaluated by both gel electrophoresis and spectrophotometric determination.2.3.cDNA synthesisThe samples were treated with amplification grade DNase I (Sigma Chem.Co,St.Louis)following the producer’s proto-col and directly used for reverse transcription.A total of 2m g of total RNA solution were incubated at80 C for 10min and then at25 C with0.01mg/ml random hexamers (Pharmacia,Biotech,Uppsala,Sweden)for10min.Subse-quently,reverse transcription was performed at42 C for1h using10U of murine myeloblastosis virus reverse transcrip-tase(Sigma Chem Co.)with1mM dNTPs in afinal volume of20m l and in the presence of20U of Ribonuclease inhibitor (Sigma Chem Co.).The enzyme inactivation was performed by heating the samples at70 C for10min.2.4.Quantification of the mRNA levels and geneamplification for EEF1A1/2Real time PCR was performed in single-plex reactions (7900/HT Sequence Detection System e Applied Biosys-tems).The specificity of the EEF1A1and EEF1A2amplicons was proved by digestion with appropriate restriction endonu-cleases.The primers used are reported in Table1.All amplifications were conducted in afinal volume of25m l of SYBR/Master Mix buffer(Applied Biosystems-Applera Corporation,USA),containing primers(300nM each), SYBR green,dNTPs(200mM each),Taq DNA polymerase and1m l of cDNA reverse transcription solution or20ng of DNA.28S rRNA,EEF1A1and EEF1A2were submitted to 40cycles of amplification with enzyme activation at50 C for2min,pre-denaturation at95 C for10min,denaturation at95 C for15s,annealing and extension at62 C for60s. EEF1A1,EEF1A2,and28S rRNA,were analyzed for each samples in a unique plate to assure the same amplification efficiency.The samples were treated with DNase prior to perform reverse transcription in order to completely exclude1545G.Grassi et al./Biochimie89(2007)1544e1552the possibility that amplicons of EEF1A could derived from non-actively transcribed retropseudogenes[26].The relative amounts of the mRNA of target genes were normalized by28S rRNA content.The copy number of DNA was calculated normalizing the DNA quantity by28S ribosomal DNA content and using the quantity of DNA of normal human liver as calibrator,assuming that it contains one copy for haploid genome.2.5.Western blottingProtein extracts were prepared from aliquots of6Â106 subconfluent(70%confluent)HepG2and JHH6cells that were washed with phosphate buffer saline solution(PBS) and lysed as described by Mansilla et al.[27]Cytoplasmic and nuclear-enriched extracts were prepared as described by Scaggiante et al.[28].Cells were resuspended in400m l/106 cells ice-cold10mM Hepes,pH7.9, 1.5mM MgCl2, 10mM KCl(solution A)containing1mM dithiotreitol, 1mM phenylmethylsulphonylfluoride,and0.05%Nonidet P-40.After10min incubation on ice,the cells were controlled by trypan blue dye incorporation to monitor membrane cell disruption and,if necessary,the incubation time was increased up to reach90%cell dye inclusion.The cells were then centrifuged at800Âg for5min at4 C,the supernatant containing the cytoplasm was collected and the pellet was rinsed once with solution A without Nonidet P-40.The cytoplasmic extract was complemented with0.5M NaCl, 25%glycerol and0.2mM EDTA(final concentrations)and stored in small aliquots atÀ80 C.The pellet containing nuclei was resuspended in80m l/106cells ice-cooled(20mM Hepes,pH7.9,420mM NaCl, 1.5mM MgCl2,0.2mM EDTA,25%glycerol(solution B)containing1mM dithiotrei-tol and1mM phenylmethylsulphonylfluoride).After20min incubation on ice with constant stirring,the suspension was vortexed for60s,then centrifuged at10,000Âg for10min at4 C.The supernatant containing nuclear-enriched extract was recovered and stored in small aliquots atÀ80 C.The protein concentration was evaluated by BCA protein assay reagent(Pierce,Rockford,IL).Western blotting was performed with20m g of total proteins separated by12%SDS-PAGE,and transferred onto a0.22m m nitrocellulose membrane(Schleicher&Schuell,Keene,NH). The antibodies used were mouse anti-eEF1A(0.5m g/ml) (Upstate Biotechnology,Lake Placid,NY),mouse anti-p27kip1 (C-19,1m g/ml),mouse anti-p16INK4(554079,2m g/ml), mouse anti-cyclin E1(554182,0.5m g/ml),mouse anti-PARP(556362,0.5m g/ml)(BD Bioscience Pharmingen, San Jose,CA),rabbit anti-actin(A2066,0.12m g/ml,Sigma-Aldrich,ST.Louis,MO).Rabbit anti-cyclin D1(M-20, 0.4m g/ml),rabbit anti-cyclin A(H-432,0.4m g/ml),mouse anti-E2F1(554213, 2.5m g/ml)and rabbit anti-GAPDH (0.2m g/ml)antibodies were purchased from Santa Cruz Biotechnology(California,Santa Cruz).After incubation, the corresponding secondary horseradish peroxidase antibodies were used(Santa Cruz Biotechnology,California, Santa Cruz)and the blots developed using an enhanced chemiluminescent substrate(Supersignal West Pico,Pierce, Rockford,IL).2.6.Apoptosis evaluationApoptosis was evaluated by the Annexin V test(Bender e Med System,Burlingame,CA)according to manufacturer’s instructions.In brief,cells were seeded at3.8Â103cells/cm2 in6wells plates and kept either in complete medium for 2.5days(70%confluent cells)or kept in complete medium overnight and then in serum free medium(starved cells)for two days.An aliquot of cells kept in complete medium was treated with1m M staurosporin(Sigma Chem.Co,St.Louis) 3h prior harvesting.The cells were then collected,resus-pended in binding buffer at the concentration of 2e5Â105cell/ml and incubated with annexin V Ig for 15min.Cells were then sedimented,resuspended in theTable1Primers sequences and ampliconsPrimers PP and AL(bp)T RD(bp)GenBank no. FwEF1A FwEF1A-RvBL5229EEF1A1mRNA EcoRV198þ30NM_001402 50-AAC ATT GTC GTC ATT GGA CA-30RvBL550-ACT TGC TGG TCT CAA ATT TC-30A-30FwEEF1A2FwEEF1A2-Rv3EEF1A2183EEF1A2mRNA SmaI126þ56NM_001958 50-GCC ACC GTC AAT AGG TGG AC-30Rv3EEF1A250-TGA TGT GGG TCT TCT CCT TG-30RvEEF1A1DNA-I FwEFIA-RvEEF1A1DNA-I151EEF1A1DNA MnlI102þ49þ39þ9J0461750-TTG ATC TTT CCC TTT CTG GT-30FwEEF1A2DNA50-ACC AAA CAT GGG GGC TTG GT-30RvEF1A2I-E FwEEF1A2DNA-RvEF1A2I-E206EEF1A2DNA PstI134þ71AF163763 50-TCC TTG CCC ATT TTG CTG GG-30Fw28SrRNA Fw28SrRNA-Rv28SrRNA8428S rRNA M1116750-TGG GAA TGC AGC CCA AAG-30Rv28SrRNA50-CCT TAC GGT ACT TGT TGA CTATGC-30PP¼primer’s pairs;AL¼amplicon length;RD¼restriction digestion,T¼target.1546G.Grassi et al./Biochimie89(2007)1544e1552binding buffer and incubated with propidium iodide for 20min at room temperature.After a final washing step,cells were re-suspended in 0.5ml of 1ÂPBS/0.5%BSA (bovin serum albumin)and analyzed by flow cytometry (FACS Canto,BD)using the DIVA software.2.7.Proliferation assaysTwelve hours before harvesting,70%confluent cells,initially seeded at 3.8Â103cells/cm 2in 6wells plates,were pulsed with bromodeoxyuridine (BrdU)at a concentration of 10m M.Cells were then prepared for BrdU staining as described [29]except that resuspension in ice cold 70%ET-OH was protracted over night,the treatment with 1M HCl 0.5%BSA was prolonged to 1h and incubation with fluorescein-isothiocyanate (FITC)e conjugated mouse mono-clonal antibody (BD PharMigen)anti BrdU was extended to 1h.Cells,resuspended in PBS containing 0.5%BSA,were analyzed by flow cytometry (FACScanto,Becton Dickinson)using the DIVA software.Senescence associated test [30]was performed between one and four days after cells reached confluence.At seeding a sterile cover glass for microscopy was placed on the bottom of a 3cm diameter well dish.At different time points,cells were rapidly washed with PBS and fixed with 2ml of Fixation Solution (2%w/vol Formaldehyde,0.2%w/vol Glutaralde-hyde in PBS)for 20min.Subsequently,cells were quickly washed twice with PBS and covered with 2ml of Staining Mixture.The staining mixture was obtained adding to a previ-ously prepared staining Solution (40mM Sodium Citrate/Orthophosphoric acid,pH 6;150mM NaCl;2mM MgCl 2)Potassium Exacianoferrate (II)and Potassium Exacianoferrate (III),to reach a final concentration of 5mM,and X-Gal (Sigma)at a final concentration of 1g/l.Staining was performed over night in the dark at 37 C.Subsequently,the supernatant was removed,cells rapidly washed twice inPBS,EEF 1A1/2 mRNA levels1.2 fold3.5 fold1234567HepG 2J HH6HepG 2J HH6E EF 1A 1 g e n e a m p l i f i c a t i o n (a r b i t r a r y u n i t s )D2.6 fold4.7 fold1234567E E F 1A 2 g e n e a m p l i f i c a t i o n (a r b i t r a r y u n i t s )EEEF1A1/2 gene amplification***EEF1A1EEF1A228 S200300400200300400100200300N e g a t i v e c o n t r o lN o r m a l l i v e r J H H 6H e p G 2H u H 7N e g a t i v e c o n t r o l N o r m a l l i v e r J H H 6H e p G 2H uH 7N e g a t i v e c o n t r o lN o r m a l l i v e r J H H 6H e p G 2H u H 7A0.511.522.5Normal liverNormal liverNormal liverNormal liverHepG 2H uH 7J HH6E EF 1A 1 m R N A l e v e l s (a r b i t r a r y u n i t s )*28-folds*24-folds*177-folds0.511.522.533.5H epG2HuH7JHH6E EF 1A 2 m R N A l e v e l s (a r b i t r a r y u n i t s )*205-folds1.5-folds2.9-foldsBCFig.1.EEF1A1and EEF1A2expression levels and gene amplification.Panel A)Non-quantitative RT-PCR analysis of the amplified genes is reported.Panel B)EEF1A1;and Panel C)EEF1A2mRNA amounts evaluated by quantitative PCR in subconfluent HepG2,JHH6,HuH7and in normal liver.Data were normalized to the quantity of 28S rRNA.Panel D)EEF1A1and Panel E)EEF1A2gene copy number evaluated in subconfluent HepG2,JHH6and in normal liver.28S rRNA was used as normalizer of the RNA and DNA quantity and normal liver DNA was used as calibrator.The results are expressed as means ÆS.D.of three independent quantifications.*P <0.05compared to normal liver tissue.1547G.Grassi et al./Biochimie 89(2007)1544e 1552then in H2O andfinally over-layered by a drop of glycerol.The cover glass wasfixed on a microscopy slide with cells turned over the glycerol drop(to avoid air bubble formation).After 30min of gentle heating at37 C to dry any excess of water, cover glasses were sealed and analysed by microscope.2.8.Statistical analysisP values were calculated using the Student’s t-test led by MS Excel.P values0.05were considered to be statistically significant.3.Results3.1.EEF1A expression levelsHepG2and JHH6cell lines are here considered models to explore the so far unknown biological role of eEF1A in hepa-tocellular carcinoma.These two cell lines have a different morphology,hepatocyte-like for HepG2and undifferentiated for JHH6[23,24].Moreover,albumin mRNA content,a known marker of hepatocyte differentiation,is more then2times more expressed in HepG2than in JHH6,further indicating the higher differentiation grade of HepG2compared to JHH6(see Section2for details).Thus,the different phenotype allows to detect possible correlation between eEF1A and the tumor cell grade.The mRNA levels of EEF1A1and EEF1A2were analyzed in sub-confluent(70%confluent)HepG2and JHH6cells.Total RNA,obtained from normal liver biopsies collected during surgical resection for angioma,was used as control. EEF1A1and EEF1A2mRNA levels were also evaluated in the HuH7hepatic cell line[23]which,compared to HepG2 and JHH6,displays an intermediate differentiation grade as evaluated by morphological inspection and albumin expres-sion(see Section2).A non-quantitative RT-PCR analysis of the amplified genes is reported in Fig.1A while the quantita-tive real time PCR data are reported in Fig.1B e pared to normal liver tissue(Fig.1B),significant(P<0.05)28,24 and177folds increase of EEF1A1mRNA were detected in HepG2,HuH7and JHH6cell line,respectively.In the case of EEF1A2(Fig.1C),a marked increase of the mRNA level (205folds)was observed in JHH6(P<0.001),while no significant increment in the mRNA levels was found in HepG2and HuH7.Based on this result,the subsequent experiments were conducted with the two most phenotypically different hepatic cell line,i.e.HepG2and JHH6.To evaluate the contribution of gene amplification to the increased mRNA levels,EEF1A1and EEF1A2genes were amplified by2specific pairs of primers.As shown in Fig.1, panels D and E,a significant(P<0.05)amplification rate of both genes was observed in JHH6compared to normal liver tissue.On the contrary,in HepG2,a significant(P¼0.05) increment was observed for EEF1A1gene only.Since a commercial antibody able to distinguish between eEF1A1and eEF1A2proteins is not available,Western blotting analysis of cytoplasmic and nuclear-enriched eEF1A content was performed.The data are reported in a representative blot in Fig.2A and summarized as nuclear-enriched/cytoplas-mic ratio in Fig.2B.An increased nuclear-enriched/cytoplas-mic ratio for eEF1A protein was observed in JHH6compared to HepG2(P¼0.038)(Fig.2B).Additionally,the tendency to have absolute higher nuclear-enriched levels of eEF1A pro-tein in JHH6compared to HepG2was observed(Fig.2A). 3.2.ApoptosisThe capacity of eEF1A1and eEF1A2to modulate programmed cell death in relation to the expression levels [16],prompted us to evaluate the apoptotic rate of HepG2 and JHH6cell lines.In sub-confluent cells(two days after cell plating in complete medium)no differences were detected among HepG2and JHH6in the apoptosis rate evaluated by the percent of annexin V positive cells(Fig.3A)and confirmed by poly-(ADP)-ribose-polymerase(PARP)cleavage(data not shown).Since the influence of eEF1A on apoptosis induced by se-rum deprivation has been reported[9,12,31],we explored the effects of serum removal on the apoptotic rate.No differ-ence was observed between JHH6and HepG2after2days of starvation in spite of a general increase in annexin V positive cells as compared to non-starved cells(Fig.3A,B).To evaluateB eEF1A nuclear-enriched/cytoplasmicprotein level0.40.81.21.62Rationuclear-enricched/cytoplasmJHH6HepG2#A eEF1A nuclear-enriched andcytoplasmic protein levelHepG2HepG2JHH6JHH6Cytoplasm Nuclear-enrichedWB:eEF1AWB :GAPDH WB :Actin--47 kDa33 kDaFig.2.eEF1A protein levels.Panel A)Representative blot showing the protein levels of total eEF1A in subconfluent JHH6and HepG2in the cytoplasm and in the nuclear enriched fraction.Panel B)The ratio between the nuclear-enriched and cytoplasm protein levels in subconfluent JHH6and HepG2is showed.The results are expressed as meansÆS.D.of three independent quan-tifications.#P¼0.038compared to HepG2.1548G.Grassi et al./Biochimie89(2007)1544e1552the effect of one additional pro-apoptic condition,cells were treated by the known apoptic inducer staurosporin [32,33].Once again,no major differences were noted in the amount of apoptotic cells (Fig.3C).Notably,in Hela cells,used as control,the amount of annexin V positive cells,i.e.apoptotic cells,did not substantially change under all the condition tested.As expected,the percent of annexin V positive cells was inversely related to the number of cells in both HepG2and JHH6cell line (Fig.3,panels D and E).3.3.ProliferationDue to the implication of eEF1A in cell proliferation [11e 14],the relation between EEF1A expression-level/sub-cellular-localization and the proliferative activity was investigated by measuring the amount of different cell cycle inhibitors (p16,p27and p21),and mediators of the cell cycle such as cyclins (cyclin E1,cyclin D,cyclin A).The amount of the transcription factor E2F1and of the newly synthesized DNA were also investigated.In sub-confluent cells,p16and p27,but not p21,are more elevated in HepG2compared to JHH6(Fig.4A).On the contrary,nosignificant difference was observed for the protein levels of cyclin E1,cyclin D1,cyclin A and of the transcription factor E2F1(data not shown).No variation was observed in the proliferative activity as evaluated by BrdU incorporation (Fig.4B).However,between two (data not shown)and four days (Fig.4C)after reaching cell confluence,HepG2exit the cell cycle more massively than JHH6cell as indicated by the evident staining for the b -gal associated senescence test.Hela cells did not significantly stain for the senescence associated b -gal test.4.DiscussionThere is a growing evidence of the involvement of compo-nents of the translation machinery in the development of cancer.In particular,the protein elongation factor eEF1A2has been identified as an important player in many human tumors [18e 20].Despite these observations,no information are so far available on its possible contribution to the biology of HCC [34].We show that the mRNA levels of both EEF1A1and 2are dramatically increased in the JHH6cells compared either toHepG2JHH6HepG2JHH6c e l l n u m b e rProliferating cells Starved cells51015202530HepG2JHH6 JHH6Hela HepG2Hela HepG2JHH6HelaProliferating cellsStarved cellsProliferating cells treatedby staurosporina p o p t o t i c c e l l s (a n n e x i n V p o s i t i v e )Fig.3.Apoptosis rate in HepG2and JHH6cells.Apoptosis rate was tested in A)subconfluent proliferating B)starved and C)subconfluent proliferating and staur-osporin treated (3h)HepG2,JHH6and Hela cells.The amount of apoptotic cells was evaluated on the basis of the %of annexin V positive cells.D)and E)show the number of cells relative to the treatments reported in A)and B).The results are expressed as means ÆS.D.of three independent evaluations.1549G.Grassi et al./Biochimie 89(2007)1544e 1552the normal liver or HepG2and HuH7cells(Fig.1B,C).Since the differentiation grade decreases from HepG2to HuH7and JHH6[23,24],it is tempting to speculate that there might be a direct correlation between the mRNA levels of eEF1A1/2 and the neoplastic phenotype of HCC cells.It is possible that a more undifferentiated HCC cell needs higher amount of eEF1A mRNA to sustain a prompt protein synthesis,possi-bly required in cell proliferation and in the organization of cy-toskeleton[6],both crucial aspects for an actively cycling/ migrating cell.Whereas only one of the two eEF1A forms is normally expressed in non-transformed cells[1]in the HCC cell lines considered,we observed the contemporary elevation of the two EEF1A mRNAs,a phenomenon particularly marked in JHH6cells(Fig.1B,C).This observation may in part depend on the parallel gene amplification detected for both eEF1A1and2(Fig.1D,E).In fact,the modest extent of amplification can hardly account for the dramatic increase of the mRNA levels observed,especially in JHH6cells.A better insight about this observation will come from the evaluation on the expression levels of eEF1A forms in an appropriate number of HCC biopsies from patients.The increased mRNA expression of EEF1A genes in JHH6 with respect to HepG2,is paralleled by an increased nuclear-enriched/cytoplasm protein ratio(Fig.2)in JHH6(P¼0.038) compared to HepG2.The fact that by means of the nuclear protein extraction procedure the cytoskeleton sedimentation occurs and with it sediments the eEF1A1bound to actin [6,35],may suggest that in JHH6,the higher levels of cyto-skeleton-bound eEF1A1are required for the non-canonical function of this protein in cytoskeletal organization[6].The higher mRNA levels detected for eEF1A1compared tonormalJHH6HepG2HeLaConfluent cells (day 0)Day 4 after reaching confluenceC-gal associated senescence testHepG2 JHH6P21P27GAPDHP16102030405060HepG2JHH6BrdUincorporationLevels of cell cycle inhibitors BrdU incorporationA BFig.4.Proliferation markers in HepG2and JHH6cells.A)Western blot of different cell cycle inhibitors performed using protein extracts from subconfluent HepG2 and JHH6cells is reported.B)Proliferation rate was evaluated measuring,byflow cytometry,the%of BrdU positive cells in subconfluent HepG2and JHH6;the results are expressed as meansÆS.D.of three independent evaluations.C)Reported are representative imagines of confluent HepG2and JHH6cells stained for senescence-associated b-galactosidase and analyzed by microscopy at a20Âmagnification.Hela cells were introduced for comparison.1550G.Grassi et al./Biochimie89(2007)1544e1552。
2018级博士生英语试卷(答)(1)

`English Final Exam for 2018 Doctoral Students(Dec. 27, 2018)Student NO.___________________ Name____________________Paper OneEnglish Writing for Biomedical PurposesPart IDirections: Choose the right one from the four choices marked A, B, C or D.1.Inconsistent with previous studies, our results from a large cohort of patients_____ this long-standing assumption.A. contrastB. compareC. reinforceD. challenge2.Patients who were receiving mechanical ventilation were considered _____ ifthey met the following modified criteria for acute lung injury or the acute respiratory distress syndrome.A. acceptableB. eligibleC. considerableD. credible3.However, results from several small studies in humans have yielded inconclusiveevidence of a beneficial _____ of ascorbic acid on lead toxicity.A. effectB. effectivenessC. affectionD. efficacy4. A _____ disease such as diabetes can affect the whole body.A. systematicB. systemicC. generalD. whole5.All tumours from AOM treated mice were _____ to histological examinationafter routine processing and haematoxylin and eosin staining.A. subjectB. subjectedC. injectedD. directed6.Serious arrhythmias are prevented whenever possible by _____ treatment ofpremonitory signs or otherwise controlled immediately after recognition byappropriate therapy.A. aggressiveB. recessiveC. abusiveD. successive7.CT scans and digital subtraction angiograms of these patients wereretrospectively reviewed by two investigators in _____ to evaluate tumor feeding vessels.A. agreementB. consentC. approvalD. consensus8.The beneficial effects of pharmacotherapy for chronic obstructive pulmonarydisease (COPD) are well _____.A. elusiveB. confirmedC. establishedD. achieved9.Chemically induced colon carcinogenesis in rodents is also suppressed by _____of NSAIDs.A. treatmentB. administrationC. managementD. registration10.Thus, it _____ further investigation of whether mfat-1 expression in diseasemodels such as non-obese mice can mitigate the development of type 1 diabetes.A. elucidatesB. interpretsC. warrantsD. guarantees11.We used a _____ questionnaire to determine whether participants met theAmerican College of Rheumatology survey criteria for gout.A. supplementaryB. complimentaryC. complementaryD. sentimental12.Ubiquitinated p53 was detected _____ immunoblotting _____ the DO-1 p53antibody.A.by...withB.for...inC.with...forD.via...on13.Cells were placed _____ a 60Co Picker unit irradiator (1.56 Gy/min) andexposed _____ 8 Gy -irradiation.B.in...withC.in...toD.on...to14.Our aim was to _____ whether or not vitamin D supplementation or deficiencyin infancy could affect occurrence of type 1 diabetes.A. studyB. ascertainC. clarifyD. research15._____ intake of purine-rich vegetables or protein is not associated with anincreased risk of gout.A. IntermediateB. ModerateC. MediumD. Immediate16.We would like to express our _____ to all the interview partners at the WorldHealth Organization for their time, expertise, and confidence.A. magnitudeB. altitudeC. aptitudeD. gratitude17.Apoptosis was analyzed _____ a FACScan(Becton Dickinson) and quantified_____ percentage of annexin-V and PI-positive.A. in...asB. on...forC. on...asD. by...for18._____ primary culture, the cells were resuspended _____ Dulbecco’s modifiedEagle’s medium containing 10% (vol/vol) fetal bovine serum and gentamicin.A.By...withB.For...inC.To...byD.At...over19.Ebola virus can spread among humans primarily through unprotected directcontact of skin or mucous membranes with blood or body fluids of a person who is ill with EVD, or the _____ of a deceased patient who had EVD.A. corpusB. corpseC. corpsD. lupus20.Treatment _____ a low dose of cadmium chloride (1 mg/kg) showed no effect onthe testis, and DAZL staining was comparable _____ control (Fig.1B).A.of...toC.at...asD.at...with21.P-gp expression was strongly induced by SJW (400% increase at 300 µg ml-1)and by HYP (700% at 3 µM) _____ a dose-dependent manner.A.onB.inC.withD.by22.Baseline ADMA levels were higher in patients who had died than in patientswho were alive at 1 year follow-up (1.23[0.98 to 1.56]_____ 0.95[0.77 to 1.20]mmol/L, p<0.001).A.fromB. B. versusC. C. toD.D. with23.The _____ for taking this approach is clear enough.A. rationaleB. notionC. hypothesisD. explanation24.This drug contains no _____ substances and has no side effects.A. toxinB. tonicC. toxicD. poisonous25.The risk of DVT and PE were significantly _____, and were highest in the firsttwo weeks, after urinary tract infection.A. roseB. raisedC. arousedD. arose26.Data was collected in the first year of life about frequency and dose of vitamin Dsupplementation and _____ of rickets.A. prescriptionB. absenceC. presentationD. presence27.Prostacyclin (PGI2) is produced from the endothelium throughcyclooxygenase-1, and binds to specific _____ in SMCs and activates adenylate cyclase.A. receiversB. receptorsC. receiptsD. recipient28.To _____ the hypothesis, experiments involving Western blots and RNAinterference were performed.A. testifyB. verifyC. justifyD. certify29.Over the past 5 decades, the proportion of DM-associated cardiovasculardiseases has been on the rise, thus _____ the need for more efforts to aggressively control the risk factors of CVDs.urgingA. urgingB. highlightingC. pressingD. enlightening30.Children _____ of having rickets during the first year of life had a RR of3.0(1.0-9.0) compared with those without the disease.A. doubtedB. suspectedC. diagnosedD. suspended31.Curcumin, a traditional medicine, exhibits anticarcinogenic andanti-inflammatory _____.A. asperityB. propertiesC. perspectivesD. prosperity32.In this study, we aimed to examine the rate of thrombolytic therapy in youngstroke patients with and without a history of migraine. We _____ that migraine would be associated with a lower rate of thrombolytic therapy.A. hypothesizesB. speculatedC. postulatedD. stipulated33.The mechanism by which PA28 exerts these effects has not been _____.A. anticipatedB. elucidatedC. remuneratedD. eliminated34.We utilized a previously described _____ to evaluate ubiquitination (Li et al,2013).A. agendaB. programC. portfolioD. protocol35.Surgical specimens of human colon cancer and adjacent normal colon mucosatissues were taken from eight Japanese patients who had _____ surgical operations for colorectal cancers at the National Cancer Center Hospital, Tokyo, and samples were immediately frozen in liquid nitrogen.A. undertakenB. undergoneC. conductedD. performed36.It consists of 10 pages of text, 2 tables, 2 pages of ____ to figures, and 6photocopies of figures.A. legendsB. accountsC. descriptionsD. introductions37.There have been no reports ____ of rosiglitazone–associated elevations in theaminotransferase level or hepatotoxicity.A. to dateB. right nowC. for nowD. to go38.As shown in Table 1, p8 was overexpressed in 71.1% of PC and in 100% of PCcell lines, ____ it was not overexpressed in MC.A. howeverB. althoughC. whereasD. albeit39.The RT-PCR assay was repeated at least three times per each sample to confirmthe ____of the results.A. reproducibilityB. availabilityC. probabilityD. likelihood40.____ asthma, Th2 cytokines are a crucial contributing factor of allergic airwayinflammation and AHR.A. In the case ofB. In case ofC. Regardless ofD. Irrespective ofPart IIDirections: Choose the right one from the given four tenses marked A, B, C or D.ResultsZebrafish nkx2.5 Can Activate myo-2 Expression When Expressed in C. elegans Body Wall Muscle.To determine whether zebrafish nkx2.5 __46__ similarly to che-22, we __47__ nkx2.5 in C. elegans Body Wall Muscle and examined expressionof the endogenous myo-2 gene by antibody staining. The rationale for this approach __48__ as follows. In wild-type C. elegans, che-22 __49__ expressed exclusively in pharyngeal muscle, whereas it __50__ expression of the pharyngeal muscle-specific myosin heavy chain gene myo-2. However, ectopic expression of che-22 in body wall muscle __51__ expression of myo-2. Because myo-2 __52__ normally never expressed in body wall muscle, this extopic expression assay provides a sensitive test for che-22 function. We __53__ two transgenic lines expressing an nkx2.5 cDNA under the control of the unc-54 body wall muscle-specific promoter. In both lines, we __54__ myo-2 expression in the body wall muscles (Fig. 1 A and B). These results __55__ that nkx2.5 can function like che-22 to induce myo-2 expression.41.A. can function B. could function C. can have functioned D. could have functioned42.A. express B. expressed C. have expressed D. had expressed43.A. was B. is C. has been D. had been44.A. is B. was C. had been D. has been45.A. activates B. activated C. has activated D. had activated46.A. could activate B. can activate C. could have activated D. can have activated47.A. was B. has been C. had been D. is48.A. generate B. have generated C. had generated D. generated49.A. detected B. detect C. have detected D. had detected50.A. showed B. show C. had shown D. have shownPart IIIDirections: Choose the one that best fits into the Discussion Section from the four choices marked A, B, C or D.DISCUSSIONThe p8 gene is barely expressed in NP but is overexpressed in acute pancreatitis (4, 12) . It is also strongly __56__ in pancreatic development and regeneration (4) . We have demonstrated that p8 is overexpressed in PC in the__57__ study. The characteristic expression of p8 is mainly attributable to its mitogenic activity (5) .__58__, p8 expression in PC would not be cancer-specific. __59__, it should be clarified whether p8 overexpression in PC is simply attributable to the excessive growth activity of cancer cells or to some genetic change(s), such as mutations.We __60__ the correlation between p8 overexpression and various clinicopathological parameters in PC. Larger tumors (>2 cm) showed a significantly higher overexpression rate of p8, and less differentiated types, advanced stages, and cases characterized by shorter survival tended to show p8 overexpression. These results also reflect the mitogenic activity of p8.__61__ reports (4, 5) have shown that p8 expression is induced by various proapoptotic __62__. It is suggested that p8 has an anti-apoptotic function (4, 5) . The significance of apoptosis in cancer cells is controversial. High spontaneous apoptosis is __63__ to be correlated with poor prognosis in PC (13) . If p8 has anti-apoptotic activity, p8 overexpression in PC cells would lead to resistance against apoptosis. Although we have not demonstrated the relationship between p8 and apoptosis in PC, the tendency toward shorter survival in p8-overexpressing cases is not __64__ with the past report (13) . It should be investigated whether p8 promotes PC cell growth through its anti-apoptotic activity.It is __65__ that p8 is a DNA-binding protein. As a transcriptional factor, it has a role in some phosphorylation/dephosphorylation signal pathways that involve its translocation to the nucleus and specific binding to DNA (4) . Potentially, p8 is phosphorylated by various kinases (4, 5) . Recent reports (14) showed that some kinases, such as the phosphatidylinositol 3-kinase or extracellular signal-regulated kinase, lead to inappropriate pancreatic cellular proliferation. Genetic mutations of K-ras, p16, and p53 in PC lead to cellular proliferation __66__ the phosphatidylinositol 3-kinase and/or the extracellular signal-regulated kinase pathways (14) . It is to be examined whether there is p8mutation in PC and how p8 participates in kinase signaling pathways.Recently, candidate of metastasis-1, a __67__ factor in human breast cancer, was identified (15) . Interestingly, p8 is structurally similar to candidate of metastasis-1 (15) . p8 might be __68__ in cancer metastasis, however, we could not find a significant difference in p8 expression between primary and metastatic lesions in our study. The relationship between p8 expression and cancer metastasis needs to be studied further.In __69__, we have demonstrated the overexpression of p8 in human pancreatic cancer. Our results suggest that p8 participates in the __70__ of pancreatic cancer, which reflects its mitogenic activity.51.A. induced B. reduced C. introduced D. seduced52.A. current B. / C. present D. former53.A. Thereafter B. Subsequently C. Additionally D. Therefore54.A. But B. Similarly C. However D. Consequently55.A. researched B. investigated C. discussed D. detected56.A. Previous B. Other C. Published D. Numerous57.A. stimuli B.stimulants C. stimulations D. simulations58.A. reported B. hypothesized C. concluded D. analyzed59.A. similar B. resilient C. consistent D. identical60.A. suggested B. confirmed C. recommended D. proposed61.A. via B. viz C. on D. along62.A. fresh B. risk C. novel D. contributing63.A. resolved B. dissolved C. immersed D. involved64.A. summarization B. summary C. end D. all65.A. attack B. onset C. development D. appearance Part IVDirections: Translate into English the Chinese phrases given in the brackets to complete the preceding sentences.1.After controlling for age, sex, race, preexisting coronary heart disease, mean arterial blood pressure,diabetes, glucose level, cholesterol level, smoking, body mass index, and study site, the presence of retinopathy____________. (与慢性心力衰竭发病危险增加2倍有关)2.Maximum mean relative enhancement ratio and mean slope of relative enhancement of lung cancerpatients____________. (明显低于健康人)3.____________ receive either alendronate (10 mg per day) or calcitriol (0.5 μg per day) a mean(±SD) of 21±11 days after transplantation. (149例病人被随机分组)4.These results establish Nrg4 as a brown fat–enriched endocrine factor ____________, includingtype 2 diabetes and nonalcoholic fatty liver disease (NAFLD). (对治疗肥胖相关疾病具有潜在作用)5.____________ reported GSPE strongly decreased NO and iNOS expression by LPS-stimulatedmacrophages. (我们的研究成果与Houde 等人之前所做的研究一致)6.Among 988 patients with gastric cancer, pernicious anemia ____________. (有11例原已确诊为恶性贫血)7.Background: Obesity____________. (被认为是结直肠癌发病的重要危险因素)8.The p8 was overexpressed (positive cells >25% in 1,000 cells) in 27 of 38 (71%) of PCs,____________. (而慢性胰腺炎中仅有17%)9.However, ____________.(几个小规模的临床研究结果没有产生充分证据证明抗坏血酸对铅毒性具有有益作用)10.____________.(使用长效β2激动剂大大改善了慢性阻塞性肺病患者的治疗效果)。
鸟的性别决定方式

鸟的性别决定方式【篇一:性别决定方式】性别决定方式性别决定的方式常见的有三种:一种是xy型性别决定,特点是雌性动物体内有两条同型的性染色体xx,雄性个体内有两条异型的性染色体xy,如哺乳动物、果蝇等。
减数分裂之后,每个配子具有一套单倍体数目的常染色体和一条性染色体。
卵子中的性染色体都是x,而在精子中性染色体可能为x,也可能为y,比例为1∶1。
精子中的性染色体决定后代性别。
在1990年,一个英国研究小组发现y染色体短布尚的sry(sex-determining region of the y)基因在男性睾丸形成过程中起关键作用,失去这个基因,个体将发育出卵巢而不是睾丸。
第二种性别决定的方式是zw型,特点是雌性动物体内有两条异型的性染色体zw,雄性个体内有两条同型的性染色体zz,如蝴蝶、鱼和鸟类等。
性别有卵子中所带有的性染色体是z还是w决定最后一种性别决定方式是xo型,o代表缺少一条性染色体,雌性具有两条x染色体(xx),而雌性只有一条x染色体,其基因型为xo雄性产生两种配子:具有一条x染色体,或者没有性染色体,精子在受精过程中决定子代的性别。
根据性别决定的原理,不论是哪种性别决定方式,后代的性别比例都是1∶1。
性别决定发生在受精的过程中,受精作用一经完成,性别也就决定了。
哺乳动物的性别主要取决于体内性染色体的组成,环境对性别的决定几乎没有影响。
但在低等一些的动物体内,如两栖类、爬行类等,性别的决定除与性染色体组成有关外,与环境的变化有一定的关系。
如青蛙等低等脊椎动物,即使性染色体组成为xy,但在温度较高的环境中也会发育成雌蛙,在温度较低的环境中,即使性染色体组成为xx,也会发育成雄蛙。
也就说低等的脊椎动物染色体对性别的决定不是很强烈的。
一些物种的性别决定缺乏性染色体,在蚂蚁和密封中,性别决定于染色体的数目,而不是性染色体,雌性由受精的卵子发育而来,是二倍体;雄性数目很少,又未受精的卵子发育而来,是单倍体。
芸薹属BoCAL和BobCAL具有促进拟南芥提前开花的功能

芸薹属BoCAL和BobCAL具有促进拟南芥提前开花的功能季必云;安颖慧;张伟;李小方【摘要】CAL编码MADS-box转录因子,参与控制花分生组织的特性,在拟南芥中发生突变时没有明显的表型;不过花椰菜的BobCAL由于终止密码子提前出现导致花球的形成,而甘蓝的BoCAL可以编码完整CAL蛋白;可见,CAL同源基因在十字花科不同种的植物中具有不同的功能,不过BobCAL是否具有促进开花功能还不清楚.此研究通过构建BoCAL和BobCAL植物双元表达载体,通过浸花法获得了转基因拟南芥,结果发现BoCAL具有促进拟南芥Col和Ws两种生态型植物提前开花的功能;而BobCAL仅能促进Col拟南芥提前开花,而在Ws生态型拟南芥中却未观察到类似的功能.【期刊名称】《上海交通大学学报(农业科学版)》【年(卷),期】2014(032)005【总页数】5页(P1-4,11)【关键词】BoCAL;BobCAL;开花;转基因;拟南芥【作者】季必云;安颖慧;张伟;李小方【作者单位】华东师范大学生命科学学院,上海200241;华东师范大学生命科学学院,上海200241;华东师范大学生命科学学院,上海200241;华东师范大学生命科学学院,上海200241【正文语种】中文【中图分类】S718146;S718149拟南芥的CAL基因和AP1基因同属MADS盒调节基因家族,具有决定花分生组织的功能。
在拟南芥中,由于AP1基因功能的冗余,cal基因突变没有明显表型,而cal 和ap1共同发生突变时,其顶端分生组织分裂能力异常强烈,最终表现出像花椰菜花球一样的形态结构。
这些遗传学资料表明CAL是一个增强AP1表型的调节子,两者共同突变时,在野生型拟南芥每朵该形成花的部位,花的决定性不能建立,仍然保留花序分生组织的特性,不断分裂,结果产生与花椰菜植株形态类似的花球结构,在拟南芥中过量表达CAL基因或AP1基因都能促进开花提前[1-4]。
TRAIL——肿瘤治疗的有效途径

TRAIL——肿瘤治疗的有效途径赵杰;章必成【期刊名称】《临床误诊误治》【年(卷),期】2012(025)004【总页数】3页(P76-78)【关键词】肿瘤坏死因子类;衔接蛋白类,信号转导;肿瘤;治疗;研究【作者】赵杰;章必成【作者单位】430070武汉,广州军区武汉总医院肿瘤科;430070武汉,广州军区武汉总医院肿瘤科【正文语种】中文【中图分类】R730.5肿瘤坏死因子相关凋亡诱导配体(tumor necrosis factor-related apoptosis-inducing ligand,TRAIL)是肿瘤坏死因子(tumor necrosis factor,TNF)超家族成员之一。
1995年Wiley等[1]首先克隆出TRAIL蛋白,与死亡因子配体(FasL)具有较高同源性,能通过与死亡受体(death receptor,DR)结合参与诱导凋亡的过程。
由于TRAIL能够选择性诱导肿瘤细胞凋亡,而对正常细胞无此作用,作为当前临床肿瘤治疗领域研究的热点,为抗肿瘤治疗提供了一个有效途径。
本文就TRAIL受体诱导的信号转导途径及其在肿瘤治疗方面的研究进展进行综述。
1 TRAIL的识别TRAIL是一种Ⅱ型跨膜蛋白,其胞外区C端有1个同源三聚体亚单位结构,能在体内特定蛋白酶的作用下从细胞表面水解形成可溶性TRAIL单体。
3个TRAIL可溶性单体与Cys230螯合的锌离子形成具有活性的同源三聚体,可与DR结合而发挥细胞毒作用。
TRAIL广泛表达于脾、胸腺、小肠、结肠、胎盘、卵巢、前列腺等多种组织细胞中,而在人脑、肝及睾丸等组织中无表达[2]。
TRAIL具有特异性诱导转化细胞、病毒感染细胞和肿瘤细胞发生凋亡的作用,而对正常细胞无影响。
此外,在激活的免疫细胞,如NK细胞、T细胞、NKT细胞、树突状细胞及巨噬细胞中也有TRAIL表达,这说明TRAIL参与了机体的免疫调节,具有维持宿主免疫及免疫稳态的功能。
碧云天Western荧光检测试剂
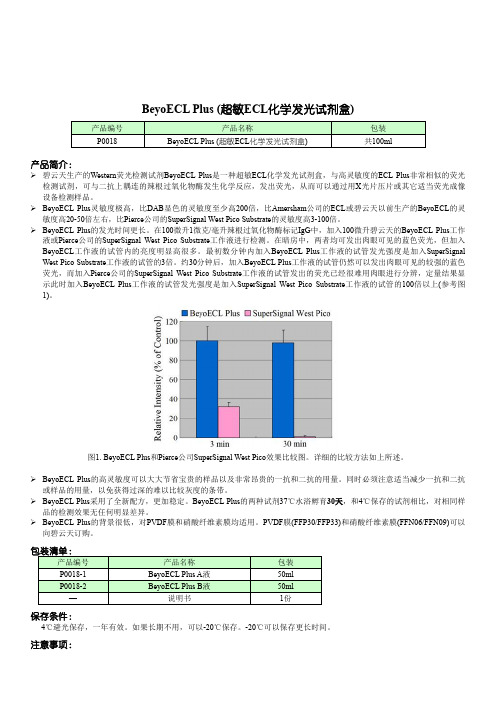
BeyoECL Plus (超敏ECL化学发光试剂盒)产品简介:碧云天生产的Western荧光检测试剂BeyoECL Plus是一种超敏ECL化学发光试剂盒,与高灵敏度的ECL Plus非常相似的荧光检测试剂,可与二抗上耦连的辣根过氧化物酶发生化学反应,发出荧光,从而可以通过用X光片压片或其它适当荧光成像设备检测样品。
BeyoECL Plus灵敏度极高,比DAB显色的灵敏度至少高200倍,比Amersham公司的ECL或碧云天以前生产的BeyoECL的灵敏度高20-50倍左右,比Pierce公司的SuperSignal West Pico Substrate的灵敏度高3-100倍。
BeyoECL Plus的发光时间更长。
在100微升1微克/毫升辣根过氧化物酶标记IgG中,加入100微升碧云天的BeyoECL Plus工作液或Pierce公司的SuperSignal West Pico Substrate工作液进行检测。
在暗房中,两者均可发出肉眼可见的蓝色荧光,但加入BeyoECL工作液的试管内的亮度明显高很多。
最初数分钟内加入BeyoECL Plus工作液的试管发光强度是加入SuperSignal West Pico Substrate工作液的试管的3倍。
约30分钟后,加入BeyoECL Plus工作液的试管仍然可以发出肉眼可见的较强的蓝色荧光,而加入Pierce公司的SuperSignal West Pico Substrate工作液的试管发出的荧光已经很难用肉眼进行分辨,定量结果显示此时加入BeyoECL Plus工作液的试管发光强度是加入SuperSignal West Pico Substrate工作液的试管的100倍以上(参考图1)。
图1. BeyoECL Plus和Pierce公司SuperSignal West Pico效果比较图。
详细的比较方法如上所述。
BeyoECL Plus的高灵敏度可以大大节省宝贵的样品以及非常昂贵的一抗和二抗的用量。
研究植物基因功能的策略和方法_3110
研究植物基因功能的策略和方法研究植物基因功能主要有两种策略:正向遗传学(forward genetics)和反向遗传学(reverse genetics)策略。
正向遗传学即通过生物个体或细胞基因组的自发突变或人工诱变,寻找相关表型或性状改变,然后通过图位克隆并结合一些基因差异表达筛选技术(如差减杂交、差异显示PCR、差异显示分析等)从这些特定性状变化的个体或细胞中找到对应的突变基因,并揭示其功能,例如遗传病基因的克隆。
反向遗传学的原理正好相反,人们首先是改变某个特定的基因或蛋白质,然后再去寻找与之有关的表型变化,例如基因剔除技术或转基因研究。
简单地说,正向遗传学是从表型变化研究基因变化,而反向遗传学则是从基因变化研究表型变化。
研究植物体内基因功能的方法主要有以下几种:(1)基因功能丧失或减少,即筛选目的基因功能部分丧失或全部丧失的突变体,比较其与野生型的表型差异来确定该基因功能;(2)基因功能增加或获得,即筛选目的基因高水平表达的植株,比较其与相应对照植株(野生型植株,功能丧失突变体或模式植物植株)差异,观察其表型性状变化来鉴定基因功能;(3)基因异位表达(Ectopic expression),通过定向调控靶基因的时空表达模式来研究基因功能;(4)微阵列(Microarray)是一种在全基因组水平对基因表达进行高通量检测的技术;(5)酵母双杂交技术(Yeast two-hybrid system)用于分析基因产物即蛋白质之间的互作。
1 基因功能丧失或减少以前,通常通过筛选自然突变体来获得基因功能部分或全部丧失的突变体,但概率较低;现在一般通过各种人工方法来获得合适突变体。
人工产生基因功能丧失的方法有插入突变、反义抑制(antisense suppression)、共抑制(cosuppression)、双链RNA干扰(double-stranded RNA interference, dsRNAi)。
Useful-expressions-of-U2-(第二单元常用表达法)
Useful-expressions-of-U2-(第二单元常用表达法)Useful expressions of U2I、Phrases:1.超过;多于;不仅仅;非常;极其2.只不过,仅仅3.不超过,至多4.与……不同5.在……中担任角色6.因为7.一些,若干,许多,大量8.纵然/即使9.与……交流10.走近,赶上,上来,被提出11.提出12.在……的尽头;在……的最后阶段;在……快要结束时13.进展;进步14.朝……走来15.发生16.偶然遇到17.来吧;赶快18.出来;出版19.以……为基础/依据20.……的数量/数目21.例如,诸如此类22.信不信由你23.利用24.与……相同25.在你的左手边26.转过拐角27.直走28.建立;增进(健康)29.目前,现在30.跟……交流31.以……为基点;在……的底部32.出席……33.眼下,暂时34.把某物交给/赠与某人35.好好/充分利用36.立即;马上;不假思索37.没有这样的事情38.由……指挥;由……控制39.控制……40.命令某人做某事41.了解/掌握……42.应某人之请求43.向某人要求某物44.要求某人做某事45.无法形容;表达不出46.表现出47.辨认出某人的声音48.把……看做……49.承认……是……50.承认……51.两个街区远的距离52.一块石头53.堵住54.封锁;封闭55.堵塞;阻碍56.官方语言57.世界经济58.发展中国家59. 西方国家60.用英语II、Sentences:1.________ ________ _______ person (不止一个人) was killed in thetraffic accident.2.__________ ______ _______ ________, he couldn’t pass the exam__________ ______ his carelessness.(信不信由你;因为)3.The novel ________ _________ _______ historical facts.(以……为依据)4.___________(然而), her son couldn’t ___________ ________(认不出她了)_________(因为)she was dirty and thin.5.He _________ ______ ________ __________ _________ German. (精通)6.Tom ___________(逐渐) ___________ ____________ ___________________(从病中康复) and returned to school.7.Our English teacher often ________ _________ ________ (利用)English songs ____________(teach) us English .8.English _______ ______ _________ _________ _____(在……中起着重要的作用) people’s life.9.________ _______ I didn’t know anybody at the party, I h ad a goodtime.(即使)10.I don’t like _____ ________ _______ ________ ________ thematter.(你处理问题方式)11.______ ________ (以……为基础)a real story, the book is popular______ __________.(目前)12.I can’t _____________ any more ideas right now.(come up/ come upwith)13.How did the problem _____________?(come up/ come up with)14.The school ____________(要求) the boy students______________(wear) long hair.15.He _______ ________ _______(迟到了) the meeting____________(由于) the bad weather.III、单选:1. Applicants(申请者)will be expected to have _______ of _______ English.A. a good command; speakB. a good command; spokenC. good commands; speakingD. good commands; spoken2. She feels strongly that each of us has a role _________ in making the earth a better place to live in.A. to have playedB. to playC. to be playedD. to be playing3. He had so much change that I could hardly _______ him.A. knowB. understandC. recognizeD. find4. Good use that the villagers have been _______ the water resources has brought them a large income.A. making ofB. putting intoC. taking onD. providing with5. The manager _______ the workers should not smoke in the office.A. askedB. toldC. requestedD. said6.—Alice, do you know what question they _______ up with at the meeting yesterday? —I have no idea.A. walkedB. cameC. cleanedD. ended7. Helen told us that she would give her another try _______ she was sure she would fail again.A. as ifB. even thoughC. unlessD. so8. He realized she was crying _________ what he had said.A. becauseB. because ofC. asD. since9. Some English programs, _______ English on Sunday, Follow Me, are very helpful to us.A. for exampleB. according toC. such asD. because of10. The famous film is ________ a Chinese fairy tale and directed by a famous director.A. basing atB. based onC. bases onD. to base atIV、短文改错:(1)Most people in the United States don’t learn to speak the second language. High schools teach languages. And very few students learn to speak good. Wh y don’t Americans speak others languages?The United States are a very large country. Americans can travel a long distance and not leave the United States. They not need to learn a second language.And many people in other parts of the world spoke English. If Americans travel to other countries, they can speak English in there, too. But some Americans think that is a mistake to speak only English. They believe it is very importantly to learn a second language.(2)When I was ten year old, I went to Hangzhou with two Americans. I became their guide. First, we went to the West Lake. They wanted to look around the lake by the boat. After buy out tickets, we got into the boat. We went around seeing the beautiful sight. After that, we went shopping. The sellers c ouldn’t spoke English. Though it was very harder, I tried my best translate the sentences. They bought some nice things under my help. And then we went to the zoo and take some pictures. When we felt hunger, we went out and had lunch. After lunch, we visited much old houses and learned more about Chinese history.。
Two basic-helix-loop-helix genes (MYC-146 and GL3) fromArabidopsis
Plant Molecular Biology52:679–688,2003.©2003Kluwer Academic Publishers.Printed in the Netherlands.679Two basic-helix-loop-helix genes(MYC-146and GL3)from Arabidopsis can activate anthocyanin biosynthesis in a white-flowered Matthiola incana mutantNicola A.Ramsay1,3,Amanda R.Walker1,4,Mark Mooney2,5and John C.Gray1,∗1Department of Plant Sciences,University of Cambridge,Downing Street,Cambridge CB23EA,UK(∗author for correspondence;e-mail jcg2@);2Genetics Department,John Innes Institute,Colney Lane, Norwich NR47UH,UK;present addresses:3Medical Research Council Laboratory of Molecular Biology,Hills Road,Cambridge CB22QH,UK;4CSIRO Plant Industry,PO Box350,Glen Osmond,SA5064,Australia;5221, Ellsworth St,San Francisco,CA94110,USAReceived11September2002;accepted in revised form7February2003Key words:anthocyanin,Arabidopsis,bHLH,Matthiola,R gene,transcription factorAbstractBasic helix-loop-helix(bHLH)proteins,similar to mammalian Myc transcription factors,regulate the anthocyanin biosynthetic pathway in both monocots and dicots.Two Arabidopsis bHLH genes,GLABRA3(GL3)and MYC-146, encode proteins that are similar throughout the predicted amino acid sequence to R and DELILA,which regulate anthocyanin production in maize and snapdragon,respectively.Northern blot analysis indicates that MYC-146is most highly expressed inflower buds andflowers.Expression of a MYC-146cDNA from the CaMV35S promoter was unable to complement the anthocyanin deficiency in a ttg1mutant of Arabidopsis and resulted in no obvious phenotypic change in Columbia plants.However,transient expression of GL3and MYC-146upon microprojectile bombardment of petals of a white-flowered mutant of Matthiola incana was able to complement anthocyanin deficiency.The lack of anthocyanin-deficient Arabidopsis mutants mapping to the locations of GL3and MYC-146 suggests that the two bHLH proteins may be partially redundant and overlap in function.Abbreviations:bHLH,basic-helix-loop-helix;GL3,GLABRA3;TT,TRANSPARENT TESTA;TTG1,TRANSPAR-ENT TESTA GLABRA1IntroductionThe anthocyanin biosynthetic pathway has been stud-ied in many plant species(Winkel-Shirley,2001). Mutants with defects in the anthocyanin pathway are viable due to the non-essential nature of the antho-cyanin pigment and are easily identified due to a lack of purple coloration.In Arabidopsis,the ma-jority of such mutants have been isolated based on altered seed-coat pigmentation and as such are known as transparent testa(tt)mutants(Koornneef,1981, 1990).Thefirst anthocyanin regulatory gene(R)was isolated from maize(Ludwig et al.,1989)and en-codes a bHLH protein belonging to a family of similar proteins(encoded by R,Lc,Sn and B)which control anthocyanin patterning in a tissue-specific manner in maize(Ludwig et al.,1990).In maize,many of the genes encoding enzymes in the anthocyanin pathway have been shown to be transcriptionally activated by bHLH and MYB proteins(Roth et al.,1991;Bodeau and Walbot,1992;Lesnick and Chandler,1998).The anthocyanin pathway is also regulated by bHLH pro-teins in dicots.DELILA controls corolla colour in snapdragonflowers(Goodrich et al.,1992),JAF13 and AN1regulateflower colour in petunia(Quat-trocchio et al.,1993,1998;Spelt et al.,2000)and TT8regulates anthocyanin synthesis in Arabidopsis seedlings(Shirley et al.,1995)and proanthocyanidin synthesis in siliques(Nesi et al.,2000).These bHLH680proteins regulate genes encoding enzymes in the latter part of the anthocyanin pathway(Martin et al.,1991; Quattrocchio et al.,1993,1998;Nesi et al.,2000).Constitutive expression of the maize Lc gene in Arabidopsis plants leads to a higher anthocyanin con-tent and an increase in the number of trichomes(Lloyd et al.,1992).Arabidopsis plants mutant at the TRANS-PARENT TESTA GLABRA1(TTG1)locus lack antho-cyanin,trichomes and seed mucilage and have ectopic root hairs(Koornneef,1981;Galway et al.,1994).All of these phenotypes are complemented by constitutive expression of Lc(Lloyd et al.,1992).However,iso-lation of the TTG1gene showed it encodes a WD40 repeat protein(Walker et al.,1999)structurally dis-tinct from bHLH proteins and involved in facilitating protein-protein interactions(Payne et al.,2000).A mutant at the G locus in Matthiola incana ex-hibits phenotypic changes similar to those shown by ttg1mutants of Arabidopsis.Mutant plants lack an-thocyanin throughout the plant body,having white flowers in contrast to the dark purpleflowers of the wild type,and also lack leaf hairs(Kappert,1949).Ex-tensive biochemical and genetic studies of this mutant suggested that the block in the anthocyanin pathway is in the regulation of genes encoding enzymes from di-hydroflavonol4-reductase onwards(Forkmann,1977; Heller et al.,1985),similar to delila and an1mu-tants(Martin et al.,1991;Quattrocchio et al.,1993). This white-flowered Matthiola mutant has been shown to be the result of a mutation in a bHLH gene lead-ing to phenotypes similar to ttg1mutations in Ara-bidopsis(N.A.Ramsay,A.R.Walker and J.C.Gray, unpublished results).The aim of this study was to identify Arabidopsis R homologues involved in regulating anthocyanin pig-mentation.To this end,we isolated two cDNAs encod-ing bHLH proteins that show extensive sequence simi-larity to R and DELILA throughout their length.Tran-sient expression of each of the cDNAs from the CaMV 35S promoter was able to complement the Matthiola G locus mutant.The pattern of expression of MYC-146, which does not map to a known transparent testa lo-cus,suggests it may regulate anthocyanin biosynthesis inflowers.However,over-expression of MYC-146had no effect on the phenotype of either Columbia eco-type plants or ttg1-9plants.One explanation is that the two bHLH genes are partially redundant and have overlapping functions.Materials and methodsPlant materialArabidopsis plant material for DNA and RNA extrac-tion and for over-expression was grown as described by Walker et al.(1999).Individual Matthiola incana plants mutant at the G locus(Kappert,1949)were grown in20cm potsfilled with Levington M3potting compost(Ipswich,UK)at22◦C in glasshouses with supplementary light and allowed toflower.All plants received a weekly feed of macronutrients(Tomorite, Levington,Ipswich,UK).Degenerate PCR and library screensDegenerate PCR primer1055(5 -AGRAGYATHCA-RTGGAGYTAYGCNATHTTYTTG-3 )was used in combination with vector-specific primer466(5 -GACTCGAGTCGACATCG-3 )to amplify fragments from aλZiplox cDNA library generated from etio-lated seedlings,roots,leaves andflowering inflores-cences(Newman et al.,1994).PCR was performed with AmpliTaq(Perkin-Elmer)under the following re-action conditions:40cycles of95◦C for1min,40◦C for2min and72◦C for3min.Products from this reaction were used as a template for the second round of PCR with nested degenerate PCR primers673(5 -CAGAGRAGYGAICARYTIMGRGARCTITAYGA-3 )and465(5 -TTYTCYCTICGYTTYCTYTCIGAY-AAIACRTGKTT-3 ).PCR conditions were as for primers1055and466,but with an annealing temper-ature of45◦C.The major PCR band of1.2kb was gel-purified,and the DNA extracted and ligated into pCRScript(Stratagene)to generate pCR3.1.A genomic library from Arabidopsis ecotype Landsberg erecta inλDASHII(Boyce et al.,1994) distributed by the EEC-BRIDGE Arabidopsis DNA Stock Centre(Köln,Germany)was screened by hy-bridisation with the insert from pCR3.1.Two clones were isolated,λCR3andλCR6;the sequences in these clones that hybridised to the PCR product were subcloned as Spe I fragments into pBluescript and analysed further.Plasmid constructionEST146D23T7was amplified in Escherichia coli XL1-Blue(Stratagene)and sequenced with an ABI 377sequencer(Perkin-Elmer,Warrington,UK).The mutated base pair identified in EST146D23T7was reverted to that of the wild-type sequence by means of681the QuikChange Site-Directed-Mutagenesis Kit(Strat-agene)to generate the plasmid pMYC-146.MYC-146 cDNA was placed under the control of the cauliflower mosaic virus(CaMV)35S promoter by isolating a Sma I-Sph I fragment from pMYC-146and ligating this into the Sma I and Sph I sites of pFF19(Timmer-mans et al.,1990),thereby producing p35S-MYC-146. p MYC-146Ox for plant transformation was generated by isolating a Sac I-Eco RI fragment from pMYC-146and ligating this with Sac I-and Eco RI-digested pROK2,a pBIN19derivative(Bevan,1985)con-taining the CaMV35S promoter and nos terminator. p MYC-146Ox was subjected to DNA sequencing to confirm the correct sequence of the inserted MYC-146 cDNA and of theflanking promoter and terminator regions of pROK2.A GL3cDNA was isolated by RT-PCR with the following primers designed from the genomic se-quence contained inλCR3:CR3Nde I(5 -GGATGAA-CATATGGCTACCGG-3 )and CR3Bam HI(5 -TTAA-CTAAGGATCCTTCAACAGAT-3 ).First-strand cDNA was synthesised from5µg of total leaf RNA by standard methods with an oligo-dT primer.The amplified GL3cDNA was ligated into the pTOPO-XL vector(Invitrogen),sequenced and PCR er-rors rectified with the Stratagene QuikChange Site-Directed-Mutagenesis Kit.This plasmid was called pCR3cDNA.The GL3cDNA sequence was removed from pCR3cDNA as a Xba I-Spe I fragment and placed under the control of the CaMV35S promoter by liga-tion into the Xba I site of pFF19(Timmermans et al., 1990),producing p35S-GL3.Northern blot analysisNorthern blot analysis was carried out as described previously(Schaffer et al.,1998).The probe specific for MYC-146was generated by PCR on pMYC-146 with the primers5 -TGGAACAACCGGGAGACTTG-3 and5 -CTGGTGTGTCCGGTAACAACT-3 .These primers amplify a320bp product between882and 1201bp relative to the start codon of AF251687. Biolistic bombardment of Matthiola petalsThe youngest open double-sterileflowers were re-moved from Matthiola incana mutant plants and placed on0.8%agar plates containing MS salts (2.13g/l).Theseflowers have reiterating petals,and the petals at the centre of theflower are still expand-ing(Lesley,1973).Fertileflowers were also used at stage5(as described by Dangelmayr et al.,1983)but the results proved to be less consistent.Gold micro-projectiles(1.6µm)were prepared as described by the supplier(BioRad).Bombardment was carried out with rupture disks of6.2MPa in a BioRad PDS-1000 Helium Particle Delivery System under a vacuum of 3.7kPa.After bombardment,flowers were incubated on plates sealed with micropore tape for1–4days at 25◦C in16h light.Plant transformationAgrobacterium tumefaciens Agl1(Lazo et al.,1991) was transformed with p MYC-146Ox by electropo-ration(Shen and Forde,1989).The construct was introduced into Columbia and ttg1-9plants by vac-uum infiltration(Bechtold et al.,1993).Transformants were selected on0.8%agar plates containing MS salts (2.13g/l)and kanamycin(35µg/ml).Kanamycin-resistant plants were screened visually for alterations in trichome number and anthocyanin content. ResultsIdentification of bHLH genes with similarity to Lc and DELILAbHLH genes potentially involved in regulating antho-cyanin biosynthesis in Arabidopsis were isolated from a cDNA library by partially nested PCR with a vector-specific primer and degenerate primers designed to conserved amino acid sequences of Lc and DELILA (see Figure1).A1247bp degenerate PCR product was amplified in the second round of PCR and encoded a protein showing61%similarity and51%identity with DELILA and58%similarity and47%identity with Lc.The PCR product was used to screen an Ara-bidopsis genomic library and two genomic clones,λCR3andλCR6,were isolated.DNA sequence analy-sis of the bHLH-encoding sequences contained in λCR3andλCR6indicated thatλCR6corresponded to the sequence of the degenerate PCR product and contained the complete genomic sequence of EST 146D23T7(accession number AF027732)located at locus At1g63650.The bHLH-encoding gene located inλCR6has been named MYC-146(Bate and Roth-stein,1997;Nesi et al.,2000),although it has also been called AtMyc-2(A.M.Lloyd and F.Zhang,data-base entry for AF027732);it will be called MYC-146 throughout this paper.λCR3contained the complete sequence of a bHLH-encoding gene located at locus682Figure1.Amino acid sequence alignment of GL3and MYC-146with DELILA and Lc.Amino acid sequence alignment of MYC-146and GL3with DELILA(from Antirrhinum;GenBank accession number M84913)and Lc(from maize;GenBank accession number M26227)both of which are involved in plant anthocyanin pigmentation.Identical amino acid residues are boxed in black and similar amino acids are boxed in grey.Dashes indicate gaps inserted to optimise alignment.The amino acid sequences used to design the degenerate primers are indicated by a bar.Intron positions in MYC-146,GL3,DELILA and Lc are denoted by triangles.The location of the HLH domain is marked by solid and dashed lines beneath the amino acid sequence.683At5g41315which,during the course of this work,was identified as GLABRA3(GL3;Payne et al.,2000). The availability of the complete Arabidopsis genome sequence(Arabidopsis Genome Initiative,2000)al-lowed the identification of139genes encoding bHLH proteins(Reichmann et al.,2000).Of these bHLH proteins,MYC-146and GL3are clearly the most similar to DELILA and JAF13,and the similarity ex-tends throughout the amino acid sequence(Figure1). Slightly lower similarity is shown to the maize R,Lc and B proteins,but the similarity extends throughout the proteins.Of the other Arabidopsis bHLH pro-teins,AtMYC1(locus At4g00480)shows the highest similarity to MYC-146and GL3,although maximum parsimony analysis indicates it is no more closely re-lated to MYC-146than R,Lc and B(Nesi et al.,2000). TT8,another Arabidopsis bHLH protein involved in regulating anthocyanin biosynthesis,is much more distantly related to MYC-146(Nesi et al.,2000).Overall,MYC-146is47%identical and60%sim-ilar to DELILA and35%identical and47%similar to R,whereas GL3is47%identical and59%similar to DELILA and36%identical and46%similar to R. MYC-146and GL3are79%identical and83%similar to each other.In addition to the bHLH motif,the pro-teins also share a highly conserved N-terminal region which,in the case of the maize B protein(encoded by a member of the R gene family),has been shown in yeast cells to be involved in protein-protein interaction with the maize MYB protein C1(Goff et al.,1992). Characterisation of the MYC-146geneThe structure of the MYC-146gene was determined by comparing the sequence of a3.5kb Spe I frag-ment fromλCR6with that of the EST146D23T7.The 3.5kb Spe I fragment contains the complete coding region of parison of the sequence ob-tained from EST146D23T7with its cognate genomic sequence inλCR6indicated that the EST contains a single C-to-T base change at position741relative to the start codon.This results in a stop codon in place of a glutamine residue at amino acid248.The MYC-146genomic sequence was also compared to the previously published genomic sequence of this gene (accession number AF013465;Bate and Rothstein, 1997).The stop codon identified in EST146D23T7 was not present in this sequence indicating that the error is in the EST.The error in the sequence of EST 146D23T7was corrected by site-directed parison of the MYC-146genomicsequence Figure2.RNA gel blot analysis of MYC-146expression.A.To-tal RNA(10µg)extracted from different organs of4-week old Arabidopsis ecotype Columbia plants was fractionated by agarose gel electrophoresis,blotted on to GeneScreen Plus and hybridised with a MYC-146gene-specific probe.The blot was stripped and then hybridised with aβ-tubulin probe as a loading control. B. Total RNA(10µg)extracted from4-week oldflowers of ttg1-13 and Arabidopsis ecotype Columbia plants was hybridised with a MYC-146gene-specific probe,as in A.The blot was stripped and then hybridised with aβ-tubulin probe as a loading control.with that published by Bate and Rothstein(1997) identified an extra intron in the MYC-146genomic se-quence(intron number6,see Figure1).The presence of this intron was confirmed by PCR amplification of this region from genomic DNA of4different ecotypes of Arabidopsis(Ws,RLD1,En1and Col,data not shown).Sequence data obtained from the Arabidopsis genome sequencing initiative provides confirmation of this intron(accession number AC011622).The posi-tions of the6introns in the MYC-146gene(Figure1) are identical to the positions of the introns in Lc and B (members of the maize R gene family;Ludwig et al., 1989;Radicella et al.,1991)and JAF13(Quattrocchio et al.,1998),consistent with a common evolutionary origin of these genes.The MYC-146gene contains an open reading frame of1788bp which encodes a protein of596 amino acid residues.The features typical of a bHLH domain are located towards the C-terminus of the pro-tein between amino acid residues405and457.These comprise a basic region of16amino acid residues, followed by helix1(16amino acid residues),a loop region of6amino acid residues and helix2(15amino acid residues;Figure1).Expression pattern of the MYC-146geneThe high level of sequence similarity between MYC-146and GL3necessitated the use of a hybridisation probe that was able to distinguish between the two684transcripts.A320bp PCR product amplified with primers to the sequence of MYC-146between882 and1201relative to the start codon was found to be specific for MYC-146(data not shown).Total RNA was extracted from different organs of Arabidopsis, fractionated by gel electrophoresis and a gel blot was hybridised with the specific32P-labelled MYC-146 probe.The MYC-146transcript was estimated to be 2.1kb and was most abundant in buds andflowers (Figure2A).To determine if expression of MYC-146 is dependent on TTG1,total RNA was extracted from wild-type and null ttg1-13plants.RNA gel blot analy-sis showed that the amount of MYC-146transcript was not decreased in the mutant plants,indicating that TTG1does not regulate the transcription of the MYC-146gene(Figure2B).Complementation of a white-flowered Matthiola mutantA white-flowered Matthiola incana mutant at the G locus(Kappert,1949)has been shown to result from a mutation in a bHLH gene similar to R and DELILA (N.A.Ramsay,A.R.Walker and J.C.Gray,unpub-lished results).Transient expression of GL3or MYC-146cDNAs following microprojectile bombardment of white mutantflowers with p35S-GL3(Figure3A)or p35S-MYC-146(Figure3B)restored the production of purple anthocyanin pigment in single cells.Bombard-ment of the whiteflowers with the vector pFF19did not produce purple spots indicating the vector alone was unable to complement the anthocyaninless pheno-type(data not shown).This result indicates that each of the two Arabidopsis genes is capable of positively regulating anthocyanin biosynthesis.Over-expression of MYC-146in Arabidopsis does not alter the phenotype of the plantConstitutive expression of the maize Lc gene in both wild-type and ttg1mutant Arabidopsis plants affected both anthocyanin and trichome production(Lloyd et al.,1992).To analyse whether over-expression of MYC-146could influence these traits,the rectified MYC-146sequence was ligated into a binary vector and used to transform ecotype Columbia and ttg1-9 mutant plants.Analysis of20transformants trans-genic for this construct did not detect any increase in trichome number or in anthocyanin content in the plant body or seed-coat of wild-type plants(data not shown).The construct also failed to complement the lack of anthocyanin in the seed coat or testa or the deficit of leaf hairs in ttg1-9mutant plants(data not shown).To confirm that the construct was being ex-pressed,RNA gel blot analysis was performed on total RNA extracted from the leaf tissue of a rep-resentative sample of T2plants,a tissue where the MYC-146transcript is not normally detected(see Figure2A).The gel blot was hybridised with the MYC-146gene-specific probe demonstrating that the amount of transcript was increased in the transgenic lines analysed(Figures4A and B),compared with the lack of hybridisation to wild-type RNA. DiscussionThefirst bHLH-motif-containing proteins,E12and E47,were identified in animals(Murre et al.,1989). Studies on these and other bHLH proteins have iden-tified the basic region as being important in facili-tating DNA binding(V oronova and Baltimore,1990) and the HLH as being responsible for homo-and hetero-dimer formation(Murre et al.,1994).bHLH regulatory proteins have also been identified in plants where the R-gene family has been demonstrated to regulate anthocyanin production in both monocotyle-donous and dicotyledonous species(Winkel-Shirley, 2001).The constitutive expression of the maize R gene in Arabidopsis suggested that,in addition to regu-lating anthocyanin production,bHLH proteins might be important regulators of several further aspects of development and gene expression including trichome formation,root hair formation and seed coat mucilage production(Lloyd et al.,1992).The regulation of these disparate pathways has been proposed to involve the interaction of TTG1(a WD40-repeat protein)with an R-like bHLH homologue and a MYB transcription factor specific for the aforementioned developmental and biosynthetic pathways(Walker and Marks,2000).In this study two Arabidopsis genes encoding pro-teins showing significant similarity throughout their deduced amino acid sequences with DELILA and Lc(known bHLH regulators of anthocyanin produc-tion;Figure1)were isolated.The availability of the Arabidopsis genome sequence(Arabidopsis Genome Initiative,2000)confirmed that these are the only two bHLH genes exhibiting such extensive similar-ity throughout their length with DELILA and Lc and identified them as possible homologues involved in regulating anthocyanin production in Arabidopsis.During the course of this work the bHLH gene lo-cated inλCR3was identified as GL3(Payne et al.,685 plementation of whiteflowers from Matthiola plants mutant at the G locus by p35S-MYC-146and p35S-GL3constructs.A. Microprojectile particle bombardment of white Matthiolaflowers with p35S-MYC-146.The petal was photographed48h after bombardment. Scale bar250µm.B.Microprojectile particle bombardment of white Matthiolaflowers with p35S-GL3.The petal was photographed48h afterbombardment.Scale bar250µm.2000).Mutations in GL3affect trichome production(Koornneef et al.,1982),lending credence to themodel that bHLH proteins are involved in regulatingthis developmental pathway in Arabidopsis.However,none of the known alleles of GL3exhibit an an-thocyanin phenotype.The MYC-146gene located inλCR6does not correspond to mutants with a knownanthocyanin phenotype,such as transparent testa mu-tants.These data suggest a number of possibilities.One is that neither GL3nor MYC-146regulate theanthocyanin biosynthetic pathway in Arabidopsis.An-other possibility is that both proteins regulate the tran-scription of the genes encoding enzymes or regulatorsof the anthocyanin pathway and that their functionsare partially redundant.In this case,a mutation in onegene would not lead to a transparent testa phenotypeif the other gene was still active.Double mutants withboth genes inactivated are very unlikely to occur in themutant plant populations screened for this phenotype.Interestingly,none of the known alleles of GL3ex-hibit a complete lack of trichomes,including the gl3-2allele which is predicted to encode a45amino acid N-terminal truncation(Payne et al.,2000),implying thatbHLH proteins able to partially complement the tri-chome phenotype do indeed exist.A candidate for thisrole would be MYC-146due to its extensive aminoacid sequence similarity with GL3(Figure1).To assess whether both MYC-146and GL3wereable to activate anthocyanin biosynthetic genes,thetwo cDNA sequences were tested for their ability tocomplement a white-flowered mutant at the G locus inMatthiola incana(a member of the Brassicaceae,as isArabidopsis).The white-flowered phenotype has beenshown to be the consequence of a mutation in a bHLH-encoding gene(N.A.Ramsay,A.R.Walker and J.C.Gray,unpublished results).Both MYC-146and GL3were equally able to activate anthocyanin biosynthesisin the Matthiola mutant(Figure3A and B).The com-plementation of the acyanic phenotype suggests thatthe function of MYC-146and GL3is partially redun-dant with respect to anthocyanin production.A secondwhite-flowered mutant of Matthiola displays a pheno-type similar to the G locus mutant(Kappert,1949).This mutation,at the E locus,results from a mutationin the Matthiola orthologue of TTG1(A.R.Walker,N.A.Ramsay and J.C.Gray,unpublished results).Nei-ther Lc,GL3or MYC-146was able to complement theanthocyaninless phenotype of the Matthiola E locusmutant.This demonstrates that,as has been observedin Arabidopsis(Koornneef,1981)and petunia(de Vet-ten et al.,1997),a WD-40repeat protein is required toactivate the anthocyanin pathway in Matthiola.The difference in expression patterns as assessedby RNA gel blot analyses indicates that MYC-146andGL3may not completely overlap in function.MYC-146expression could be detected only inflower budsandflowers(Figure2A),whereas GL3expression(detected with a GL3-specific probe)was observed686Figure4.RNA gel blot analysis of transgenic Arabidopsis plants transformed with p MYC-146Ox.A.Total RNA(10µg)was ex-tracted from the leaves of Columbia plants transformed with p MYC-146Ox(lanes1–6)and of an untransformed Columbia plant (lane7).The RNA gel blot was hybridised with a MYC-146 gene-specific probe.The blot was then stripped and hybridised with aβ-tubulin probe as a loading control.B.Total RNA(10µg)was extracted from the leaves of ttg1-9mutant plants transformed with p MYC-146Ox(lanes1–4)and of an untransformed ttg1-9plant(lane 5).The RNA gel blot was hybridised with a MYC-146gene-specific probe.The blot was then stripped and hybridised with aβ-tubulin probe as a loading control.in roots,leaves,stems,cauline leaves,flower buds andflowers(data not shown).It is possible,how-ever,that the expression pattern of MYC-146may overlap more extensively with that of GL3under con-ditions known to induce anthocyanin synthesis;this remains to be investigated.Inflowers and siliques, MYC-146may interact with TTG1and a MYB pro-tein,possibly PAP1(Borevitz et al.,2000)or TT2 (Nesi et al.,2001),to control the expression of an-thocyanin biosynthetic genes.TT8encodes a bHLH transcription factor,more distantly related to known regulators of anthocyanin biosynthesis,whose expres-sion in siliques and young seedlings is dependent on the presence of TTG1(Nesi et al.,2000).Unlike TT8, MYC-146expression is not dependent on the presence of TTG1(Figure2B)and neither is expression of GL3(Payne et al.,2000).It is possible that a transcription complex comprising TTG1,MYC-146or GL3and a MYB transcription factor may activate expression of TT8.TT8has been shown to control the expression of DFR and BANYULS but not LDOX(leucoanthocyani-din dioxygenase;Nesi et al.,2000).A further role for MYC-146and GL3might be to control expression of the genes in the latter half of the anthocyanin pathway, such as LDOX,which are unaffected by TT8.Unpub-lished results described in Nesi et al.(2000)suggest that the MYC-146transcript can be detected in silique tissue lending support to a model where MYC-146and TTG1interact to activate TT8transcription.Over-expression studies in wild-type and ttg1-9 mutant Arabidopsis plants did not provide evidence that MYC-146regulates anthocyanin or trichome pro-duction despite the fact that increased levels of tran-script could be detected(Figure4A and B).This suggests that in wild-type plants the activity of MYC-146may be limited by the availability of TTG1or MYB transcription factors.The results of these over-expression studies are in contrast to over-expression of a full-length genomic GL3sequence in wild-type and ttg1-1mutant plants which showed an increase in trichome numbers and a slight increase in anthocyanin (Payne et al.,2000).Payne et al.,(2000)also cited unpublished results indicating that over-expression of EST146D23T7was able to complement ttg1-1and to cause an overproduction of trichomes in wild-type plants;this report is difficult to reconcile with our results.AcknowledgementsWe are extremely grateful to Sue Aspinall for skilled technical assistance and to Vera Hemleben(Univer-sity of Tübingen)for supplying Matthiola incana seed. EST146D23T7was provided by the Ohio Stock Cen-tre.The work was supported by BBSRC ROPA award 9708825and BBSRC research grant G08606to J.C.G. ReferencesArabidopsis Genome Initiative.2000.Analysis of the genome se-quence of theflowering plant Arabidopsis thaliana.Nature408: 796–815.Bate,N.J.and Rothstein,S.J.1997.An Arabidopsis Myc-Like gene (MYC-146)with homology to the anthocyanin regulatory gene Delila(Accession No.AF013465).Plant Physiol.115:315. Bechtold,N.,Ellis,J.and Pelletier,G.1993.In-planta Agrobacterium-mediated gene-transfer by infiltration of adult。
biol reprod
BIOLOGY OF REPRODUCTION(2013)89(4):78,1–11Published online before print14August2013.DOI10.1095/biolreprod.113.110957A MicroRNA(mmu-miR-124)Prevents Sox9Expression in Developing Mouse Ovarian Cells1Francisca M.Real,3Ryohei Sekido,4Darı´o G.Lupia´n˜ez,3Robin Lovell-Badge,4Rafael Jime´nez,2,3and Miguel Burgos33Departamento de Gene´tica e Instituto de Biotecnologı´a,Universidad de Granada,Centro de Investigacio´n Biome´dica, Granada,Spain4Division of Stem Cell Biology and Developmental Genetics,Medical Research Council National Institute for Medical Research,Mill Hill,London,United KingdomABSTRACTIn mammals,sex differentiation depends on gonad develop-ment,which is controlled by two groups of sex-determining genes that promote one gonadal sex and antagonize the oppositeone.SOX9plays a key role during testis development in all studied vertebrates,whereas it is kept inactive in the XX gonad at the critical time of sex determination,otherwise,ovary-to-testis gonadal sex reversal occurs.However,molecular mech-anisms underlying repression of Sox9at the beginning of ovarian development,as well as other important aspects of gonad organogenesis,remain largely unknown.Because there is indirect evidence that micro-RNAs(miRNA)are necessary for testicular function,the possible involvement of miRNAs in mammalian sex determination deserved further ing microarray technology,we have identified22miRNAs showing sex-specific expression in the developing gonads during the critical period of sex determination.Bioinformatics analyses led to the identification of miR-124as the candidate gene for ovarian development.We knocked down or overexpressed miR-124in primary gonadal cell cultures and observed that miR-124 is sufficient to induce the repression of both SOX9translation and transcription in ovarian cells.Our results provide the first evidence of the involvement of a miRNA in the regulation of the gene controlling gonad development and sex determination.The miRNA microarray data reported here will help promote further research in this field,to unravel the role of other miRNAs in the genetic control of mammalian sex determination.cell transfection,gene expression,gonad development, microarray,microRNA,miR-124,ovary,regulation of translation, sex determinationINTRODUCTIONThe process of sex determination involves mechanisms that prompt the undifferentiated embryonic gonad to follow either of the two alternative developmental pathways,the testis or the ovary.A single gene located on the Y chromosome,the sex-determining region of the Y(SRY)gene,is the switch for sex determination in almost all mammals[1–3].In XY mice, shortly after the expression of Sry in somatic cells of the supporting cell lineage of the gonad,testis development begins with the differentiation of these cells as pre-Sertoli cells located in the testis cords that also enclose the primordial germ cells. Several additional cellular events also occur outside the testis cords,including mesonephric cell migration,testis-specific vascularization,and differentiation of Leydig and peritubular myoid cells[see refs.4and5for reviews].In females,the genetic pathways for ovary development are largely unknown to date.Many genes are currently known to be involved in gonadal development[6–8].Expression of SRY-related HMG-box, gene9(SOX9)is essential for normal testis development in most vertebrates.Ectopic expression of Sox9in the gonads of XX mice leads to a female-to-male sex reversal with the formation of XX testes[9,10],and XY mice lacking Sox9 expression develop ovaries,thus showing a male-to-female sex reversal[11,12].Hence,Sox9,like Sry,is necessary and sufficient to trigger testis organogenesis.A positive feedback loop between Sox9and fibroblast growth factor9(Fgf9)is initiated by Sry,which results in the up-regulation of Fgf9and the repression of wingless-type MMTV integration site family, member4(Wnt4)in the male gonad[13].This is crucial for the maintenance of testis development,because Wnt4represses the migration of endothelial and steroidogenic cells from the mesonephros to the XX gonad[14],probably by activating the b-catenin signaling pathway.The fact that XY Fgf9/Wnt4 double mutants developed testes indicates that the primary role of Fgf9is the repression of Wnt4and suggests that the Sox9/ Fgf9positive feedback loop is established through Wnt4,thus Fgf9repressing a Sox9repressor[15].Ovarian differentiation was assumed to be the default pathway in gonad development.However,several discoveries have shown that the ovarian phenotype,once determined,must be actively maintained throughout life.Although Foxl2is required to maintain the ovarian phenotype at the postnatal stage[16],little is known on how ovary differentiation is initiated at the embryonic stage after sex determination.Sex determination is established in terms of a balance between Fgf9-and Wnt4-expression predominance.It is known that Sry tips the balance toward the testis pathway in males,and Wnt signaling may have a similar role in females.R-spondin family member1(RSPO1),which,like Wnt4,serves to stabilize b-catenin,appears sufficient to block the testicular pathway in some XX humans[17],although this is not the case in mice, where XX Rspo1null mutant gonads have a phenotype similar1Supported by the Andalussian Gobernment,Junta de Andalucı´a, through grants CVI-2057and BIO-109,by the Spanish Ministry of Science and Innovation through grant CGL2011-23368,and by the UK Medical Research Council(U117512772).2Correspondence:Rafael Jime´nez,Universidad de Granada,Genetica Facultad de Ciencias Campus Fuentenueva,Granada,GR Spain18071. E-mail:rjimenez@ugr.esReceived:21May2013.First decision:31May2013.Accepted:5August2013.Ó2013by the Society for the Study of Reproduction,Inc.This is an Open Access article,freely available through Biology of Reproduction’s Authors’Choice option.eISSN:1529-7268ISSN:0006-3363to that of Wnt4null mutants[18].Available data suggest that Rspo1may cooperate with Wnt4to stabilize b-catenin,helping to counteract the establishment of the Sox9/Fgf9positive feedback loop during mouse ovarian development(see reference[19]for a review).Indeed,mice carrying a constitutive active form of b-catenin show XY female sex reversal[20].Before sex determination,Sox9expression occurs at a basal level in both male and female gonadal primordia,but at11.5 days postcoitum(dpc),it is up-regulated in males and down-regulated in females[21].The regulatory action of SRY on Sox9has recently been unraveled.SRY,in a synergistic action with NR5A1/SF1,up-regulates Sox9in male undifferentiated gonads by binding to a testis-specific Sox9enhancer[22]. However,it is not known how Sox9is down-regulated in the female gonad.Although Wnt4null mutant XX gonads can show transient upregulation of Sox9expression[13],null mutations in either the gene for b-catenin(Ctnnb1)or Foxl2are not sufficient to maintain de-repression of Sox9expression in the early XX gonad and neither are compound null mutations involving Wnt4and Foxl2,or Rspo1and Foxl2[23,24](see also our unpublished data).All this suggests that additional genetic elements other than those currently known are involved in sex determination,especially those required to drive ovary and repress testis differentiation.In this context,several studies have suggested that micro-RNAs(miRNAs)are involved in this process,but this hypothesis has not yet been sufficiently explored.MicroRNAs are small,noncoding RNAs that regulate the expression of target genes,either by guiding the cleavage of their target mRNAs or by inhibiting their translation[25–28]. miRNAs have been shown to exert post-transcriptional control of genes involved in development and other biological aspects including disease,cell death,cell proliferation and hematopoi-esis,in a variety of organisms,including Caenorhabditis elegans,flies,mammals and plants[26,29–34].Although no miRNA is currently known to play a role in vertebrate sex determination,there is indirect evidence to suggest that miRNA could be involved in the process:several miRNAs show a sexually dimorphic expression pattern in the mouse gonad at 13.5dpc[35];and targeted disruption of Dicer1,a gene involved in miRNA maturation,has revealed that Dicer protein is necessary to maintain Sertoli cell function[36–38].MATERIALS AND METHODSAnimals and Gonadal CellsGonads were dissected from Swiss,Parkes,and CD1outbred mouse embryos at11.5and13.5dpc and were either fixed or prepared for cell culture. Individual embryos were sexed either by gonad morphology(at13.5dpc)or at earlier indifferent stages using sex chromatin staining of amniotic cells[39]and duplex PCR for zinc finger protein1,Y linked(Zfy1)and the gene for myogenin(Myog).For purification of the RNA used in microarray experiments, a total of24male11.5dpc,19male13.5dpc,31female11.5dpc,and18 female13.5dpc gonads were unambiguously identified,pooled,and processed (see later discussion).For transfection studies,sexed male and female gonads were pooled,disaggregated both mechanically and enzymatically(125l g/ml collagenase;Roche)and cultured either in LabTek chamber slides or in24-well plates,using Dulbecco modified Eagle medium(DMEM;Sigma)with10% fetal calf serum(FCS,Sigma).Similarly,primary chondrocyte cultures were from mouse embryonic limbs.For in situ hybridization,sexed gonads were fixed overnight in4%paraformaldehyde and further immersed in30%sucrose (w/v),embedded in Tissue-Tek optimal cutting temperature compound (Sakura)and sectioned in a cryostat.Mouse housing and handling,as well as laboratory protocols,were approved by the University of Granada Ethics Committee for Animal Experimentation or were carried out under a U.K.Home Office Project License.MicroRNA Profiling of Mouse GonadsTotal RNA from embryonic gonads at11.5and13.5dpc was purified using a miRNeasy mini kit(Qiagen)according to the manufacturer’s protocol. Further steps were performed by Exiqon miRNA Profiling Service.The quality and integrity of the RNA samples was assessed in a Bioanalyser2100(Agilent Technologies),and the concentration was measured with a spectrophotometer (Nanodrop).All samples used for array hybridization showed RNA integrity numbers higher than9.Micro-RNA samples were labeled using a miRCURY Hy3/Hy5power labeling kit from Exiquon.Four embryo gonad samples were analyzed:male11.5dpc,female11.5dpc,male13.5dpc,and female13.5dpc. Each sample was labeled with Hy3and double hybridized against a pool of the four samples labeled with Hy5,which was used as a common reference. Hybridizations were performed with an HS400/4800hybridization station (Exiqon-Tecan).The miRCURY locked nucleic acid(LNA)microarray slides were scanned using a G2565BA microarray scanner system(resolution10l m; Agilent Technologies).Array Slide Quality Control Using Spike-InsHy3and Hy5labeling reactions,hybridization,and performance of array experiment were evaluated with RNA controls(spike-in)added at various concentrations,covering the full signal range,to the labeling reactions.A high correlation for both the Hy3and the Hy5channels indicated that both labeling and hybridization were successful.Each spike-in control had32replicates of capture probes on the array.MicroRNA Microarray Data ValidationData from selected micro-RNAs were validated in our laboratory by real-time RT-PCR using the miRCURY LNA micro-RNA PCR system(Exiqon),in a Chromo4real-time thermocycler(Bio-Rad)according to Exiqon protocols. As primer sets specific for the amplification of miR-124were not available from Exiqon,microarray data from this micro-RNA were validated using TaqMan micro-RNA assays from Applied Biosystems,according to the manufacturer’s procedures.For miR-124,three gene expression quantification experiments were performed with different gonadal RNA samples,and each quantitative(Q)-RT-PCR assay was done in triplicate using the U6snRNA as a reference.MIAME-compliant data were submitted to ArrayExpress accession: E-MEXP-2252,experiment name:Expression of miRNAs in mouse embryo gonad during the critical period of sex determination;specified release date: May11,2013.Identification of Active miR-124GenesRT-PCR was used to identify which of the three possible miR-124 precursor transcripts were expressed in the developing gonads.As the predicted amplification products are short sequences,short oligonucleotides(oligos)were designed with the eprimer3application from the emboss package,and the alignment temperature of the oligos was increased and adjusted,adding unspecific nucleotides at the50end of each oligo.The primers sequences were as follow:forward50-GCC TCT CTC TCC GTG T-30and reverse50-CCA TTC TTG GCA TTC A-30for mmu-miR-124-1;forward50-AGA GAC TCT GCT CTC CGT GT-30and reverse50-CTC CGC TCT TGG CAT TC-30for mmu-mir-124-2;and forward50-GGC TGC GTG TTC ACA G-30and reverse 50-ATC CCG CGT GCC TTA-30for mmu-mir-124-3.One triplicate quantitative(Q)-RT-PCR reaction was carried out using RNA samples from six pooled13.5dpc mouse embryonic gonads of each sex. BioinformaticsMicroarray image analysis was carried out using ImaGene version7.0 software(BioDiscovery,Inc.).The quantified signals(background correction Normexp,with offset value10[40]were normalized by Exiquon miRNA Profiling Service,using the global LOcally WEighted Scatterplot Smoothing (LOWESS)regression algorithm.In order to assess the biological significance of differentially expressed miRNAs,we searched for putative targets in genes involved in sex determination or gonad development,fetching miRNA data from miRbase(/),gene descriptions from Ensembl (/),and filtering the results with Gene Ontology(GO) terms(/)by using Perl scripts(http://www.perl. org/)developed for this purpose.Graphs were drawn and statistics were analyzed with gnu-R software(/).REAL ET AL.Luciferase AssaysHEK293T cells cultured in24-well plates were cotransfected using Lipofectamine2000(Invitrogen),with a miR-124overexpression plasmid vector(1l g/well;ref.MmiR3282-MR04;Genecopoeia),and pLuc-Sox930-untranslated region(UTR)(0.2l g/well).Forty-eight hours after transfection, cells were harvested for luciferase assay.Dual reporter assays were performed by measuring both firefly and Renilla luciferase activities(LucPair miR dual luciferase assay kit;ref.LPFR-M010;Genecopoeia)according to the manufacturer’s protocols.MicroRNA In Situ HybridizationIn situ hybridization experiments with a miR-124-specific miRCURY LNA micro-RNA detection probe(catalog no.33007-01;sequence:CTTGGCATT CACCGCGTGCCTTA;provided at25l M;Exiqon)were performed.Briefly, 8-l m-long cryosections(four per slide)were hybridized with30nM50-digoxigenin-labeled LNA probe overnight at588C.Signals were revealed using anti-digoxigenin alkaline phosphatase-conjugated antibodies(1:1500 dilution;Roche)and bone morphogenic protein(BMP)purple substrate.In situ hybridization experiments were performed six times with different gonad samples.For negative controls,gonad cryosections were subjected to the entire protocol but the probe was omitted from the hybridization mixture. MicroRNA Knock-Down and Overexpression in Cultured CellsPrimary gonadal cell cultures were transfected for miRNA inhibition and overexpression experiments.The inhibition of miR-124in XX embryonic gonad cells was produced with a high-affinity LNA-enhanced probe (antagomir)for specific in vitro knock-down(ref.139452-04;miRCURY LNA micro-RNA inhibitor50-fluorescein-labeled;from Exiqon).As a negative control,we transfected the same type of cells with a different antagomir, specific for miR-144,an miRNA that is not expressed in the female gonad according to our miRNA microarray data.Cells were plated to a density of 50000cells per cm2in8-well chamber slides(LabTek)for immunofluores-cence studies or in24-well culture plates for further RNA extraction,in500l l of DMEM supplemented with10%FCS.For transfection with the silencing probes,cells were transfected for4–6h after plating with a mixture of1.5l l of Lipofectamine2000(Invitrogen)in50l l of Opti-Mem-I reduced serum medium without antibiotics and6l l of probe in50l l of Opti-Mem,according to the guidelines of the manufacturer.After24h,the medium was replaced with DMEM supplemented with10%FCS and15l g/ml tetracycline.Forty-eight hours later the efficiency of transfection was checked by the fluorescence of the probe.The efficiency of transfection was higher than90%in all these experiments.Seventy-two hours after transfection,the cells were fixed in4% formaldehyde or processed for total RNA extraction.Overexpression of miR-124was performed using a precursor miRNA expression clone for the mouse mmu-mir-124-2gene(ref.MmiR3282-MR04; Genecopoeia).In this case,cells transfected with a precursor miRNA-scrambled control clone(ref.CmiR0001-MR04;Genecopoeia)were used as the negative control.For transfection of primary cultures of embryonic gonad cells with these vectors,we followed a procedure similar to that described previously with some exceptions.Before culture,cells were enzymatically disaggregated with collagenase(250l g/ml)and then transfected with1l g of vector by using an electroporator(Nucleofector II;Amaxa),obtaining a transfection efficiency higher than60%,which was determined by counting the percentage of fluorescent cells(green fluorescent protein[GFP]was included in the transfection vector).Forty-eight hours after transfections,cells were either fixed in4%formaldehyde or processed for total RNA extraction.Both knock-down and overexpression experiments were performed more than five times using3–413.5dpc pregnant females per experiment,providing approximately60embryonic gonads,which were divided into three sets of20 gonads each.Cells from the first set were treated with the miR-124-antagomir/ overexpression vector,those from the second set were treated with the miR-144-antagomir/control vector,and those from the third set remained untreated before fixation or RNA extraction.Gene Expression QuantificationTo check the genetic effects of the miRNA knock-down experiments by Q-RT-PCR,total RNA was extracted with Trizol reagent(Qiagen)after transfection and300–900ng total RNA was reverse transcribed using Superscript II(Invitrogen)and random hexamers as primers.RNA samples were purified from both nontransfected and transfected cells with the miR-124-silencing LNA probe.Expression levels of mmu-miR-124were quantified using TaqMan micro-RNA assays(Applied Biosystems)according to the manufacturer’s procedures.Quantification of Sox9expression levels was performed using1-l l aliquots of a1:10dilution of the RT reaction as templates in the Q-PCR reactions.Each PCR reaction was run in triplicate,and all experiments were repeated at least three times.All samples were normalized relative to glyceraldehyde-3-phosphate dehydrogenase(Gapdh),using the 2–DD CT(Livak)method.The primers for Sox9mRNA amplification were forward50-CGG AGG AAG TCG GTG AAG A-30and reverse50-GTC GGT TTT GGG AGT GGT G-30.For Gapdh amplification the primers were: forward50-GGC ATT GCT CTC AAT GAC AA-30and reverse50-TGT GAGG GAG ATG CTC AGT G-30.The identity of the amplified fragments was confirmed by sequencing in both cases.SOX9Protein DetectionImmunofluorescence analyses were performed to check the presence of SOX9protein in transfected gonadal cells.Cells were cultured in LabTek chamber slides,fixed for5min in4%formaldehyde,washed in PBS,blocked for1h at room temperature with2%BSA in PBS,and incubated overnight with a1:100dilution of SOX9-specific rabbit primary antibody(two different antibodies were used,antibody code sc-20095[Santa Cruz Biotechnology]and code AF3075[R&D Systems],and the same results were obtained).Slides were then washed in PBS and incubated for1h at room temperature with a1:500 dilution of Alexa Fluor555goat anti-rabbit secondary antibody(Invitrogen). Sections were then washed in PBS,incubated in a solution of100ng/ml40,6-diamidino-2-phenylindole(DAPI)for30min at room temperature,washed again in PBS,and mounted(Vectashield mounting medium;Vector Laboratories).Images were obtained with an Olympus BX41microscope with epifluorescence equipment.RESULTSMicroRNA Expression Profiling of Mouse11.5-and13.5-dpc Male and Female GonadsAll data from miRNA microarray profiling experiments in the present study are provided in Supplemental Data(Project-Summary.xls;all Supplemental Data are available online at ).Among757miRNAs included in the miRCURY LNA microarray slides,71miRNAs showed differential expression either between sexes or between developmental stages.The expression profiles of these miRNAs are shown in a heat map diagram(Fig.1),as previously described[41],where rows represent miRNAs and columns represent different samples.This diagram also includes a two-way hierarchical clustering of genes and samples.The miRNA clustering tree,constructed based on log2(Hy3:Hy5)ratios(.0.5SD among the four samples), permits identification of five different gene clusters(Fig.1,A–E).The miRNAs in clusters A and E are either down-regulated (Fig.1A)or up-regulated(Fig.1E)during the transition between the two developmental stages(11.5and13.5dpc)but show no great differences in levels of expression between testes and ovaries at13.5dpc.Therefore,these miRNAs are probably not involved in gonadal sex differentiation and thus have little relevance to the current study.In contrast,miRNAs in clusters B,C,and D,show differential expression in a sex-specific fashion at13.5dpc but not at11.5dpc and are therefore good candidates to be involved in gonadal sex differentiation.Cluster B includes3miRNAs that are up-regulated in ovaries and down-regulated in testes at13.5dpc, suggesting a potential role in ovarian development.Cluster C contains six miRNAs that are exclusively up-regulated in13.5 dpc testes,suggesting a role in testis development.On the other hand,cluster D consists of11miRNAs up-regulated only in 13.5dpc ovaries and,therefore,similar to those in cluster B; these miRNAs could be involved in ovarian development.The outcome of our miRNA microarray profiling experi-ments was validated by Q-RT-PCR.The expression levels of at least2representative miRNAs of each cluster,except formiR-124REGULATION OF SOX9IN OVARIAN CELLScluster E,were measured and compared with the corresponding array expression profiles (Fig.2A and Supplemental Fig.S1).In general,relative expression levels in microarrays correspond to those in the Q-RT-PCR assays.Although mmu-miR-126-5p and mmu-miR-103were classified as members of cluster A by the clustering analysis,because differences in expression levels between developmental stages are higher than those between sexes at 13.5dpc,the differential expression in 13.5dpc male and female gonads should be taken into account.Thus,we included these miRNAs in the Q-RT-PCR validation set.Our results confirmed that the expression of both miRNAs is significantly higher in females than in males at 13.5dpc (miR-103also at 11.5dpc),suggesting a potential role in ovarian development.Identification of mmu-mir-124as a Candidate Gene for Ovarian DevelopmentWe performed bioinformatic analyses to investigate the potential biological role of miRNAs that showed sex-specific expression and thus to identify new genetic elements involved in mammalian sex determination.We searched mainly specific miRNA target sequences in the 30-UTR regions of genes associated with GO terms such as,sex determination ,sex differentiation ,gonad differentiation ,testis development ,ovary development ,gonad development ,Sertoli cell differentiation ,and follicle cell differentiation .Using these criteria,we identified miR-124as a good candidate gene for ovarian development because several genes involved in sex determi-nation,including Sox9,share potential targets of this miRNA in their 30-UTR regions.Furthermore,miR-124was included in cluster B (differentially expressed in developing ovaries)and is known to control Sox9expression in the brain [42].These data strongly suggested that miR-124may play an important role in early steps of ovarian development,and thus,we carried out further analyses of this miRNA.The functional interaction between miR-124and Sox9was examined in the luciferase reporter assays.We found that miR-124significantly reduced the activity of the luciferase gene fused to the Sox930UTR by more than 70%,supporting the notion that miR-124can modulate Sox9expression by binding to its 30-UTR (Supplemental Fig.S2).Cheng et al.[42]previously described a similar effect by using a different reporter vector.miR-124is Up-Regulated in the Mouse Female Gonad Between 11.5and 13.5dpcIn order to confirm the expression data reported by our microarray screening for miR-124,we also performed Q-RT-PCR analyses and in situ hybridization with mouse embryonic gonads.Expression quantification by Q-RT-PCR confirmed that miR-124is up-regulated in the ovary but not in the testis during the critical period of gonad differentiation between 11.5and 13.5dpc (Fig.2A).According to miRBase,there are three predicted precursor hairpin sequences of mmu-miR-124in the mouse genome,giving rise to the same mature sequence.The identity of the miR-124precursor(s)expressed in mouse embryonic gonads was investigated by Q-RT-PCR with sets of primers specific for the three miR-124pre-miRNAs.Our results revealed that all three miR-124genes are expressed in 13.5dpc mouse ovaries (mainly pre-miR-124-2)but not in testes of the same developmental stage,where pre-miR-124-1is absent and the other two precursors show low expression levels (Fig.2B).Expression values were always significantly higher in females than in males for the three precursors (P¼FIG. 1.Heat map diagram showing miRNA expression profiles in embryonic gonads during the period of sex determination.A two-way hierarchical clustering of genes and samples identified five different gene clusters.Clusters A and E include miRNAs showing no sex-specific down-regulation (A)or up-regulation (E)during the transition between 11.5and 13.5dpc developmental stages;these genes are thus probably not involved in sex determination.Cluster B contains three miRNAs up-regulated in 13.5-dpc ovaries and down-regulated in 13.5testes,with a potential role in ovarian development.Cluster C includes 6miRNAs up-regulated in 13.5dpc testes but not in 13.5dpc ovaries,with a possible role in testis development.Cluster D contains 11miRNAs up-regulated in 13.5dpc ovaries but not in 13.5dpc testes,suggesting a role in ovarian development.REAL ET AL.FIG.2.Spatiotemporal expression pattern of miR-124in mouse embryonic gonads during the period of sex determination.A )Q-RT -PCR (gray bars)and miRNA microarray (white bars)data of miR-124expression levels in male (M)and female (F)gonads at 11.5and 13.5dpc.miR-124is sex-specifically up-regulated in the 13.5dpc female gonad.B )Q-RT-PCR quantification of the active miR-124precursors 1,2,and 3,in male (gray bars)and female (white bars)embryonic gonads.Amplified products derived from all three precursors are mainly detected in the female samples.C –F )In situ hybridization detection of miR-124in developing mouse gonads.Hybridization signal is very weak in 11.5dpc gonads of both sexes (C and E )and becomes prominent in the coelomic epithelium of the XY gonad and the cortical region of the 13.5dpc XX gonad (D and F ,arrows in photomicrographs).G,gonad;M,mesonephros .Dashed lines mark the border between the gonad and the mesonephros.Bar ¼300l m for all images.miR-124REGULATION OF SOX9IN OVARIAN CELLS0.015for pre-miR-124-1;P¼0.016for pre-miR-124-2;P¼0.010for pre-miR-124-3).In situ hybridization with a miR-124-specific probe showed a faint or very weak expression inboth male and female gonads at11.5dpc(Fig.2,C and E).At 13.5dpc,miR-124expression was prominently up-regulated inthe cortical region of the female gonad,whereas it wassustained,albeit weak,in the coelomic epithelium of the male gonad(Fig.2,D and F).Knock-Down of miR-124Induces Up-Regulation of Sox9in 13.5-dpc Embryonic XX Gonadal CellsBecause the30UTR region of Sox9contains a miR-124 target sequence,we hypothesize that miR-124could be responsible for maintaining down-regulation of Sox9in the female gonad from the sex differentiation stage onward.To test this hypothesis,we transfected13.5dpc embryonic gonadal cells with a specific miR-124-silencing probe,designated antagomir-124or Ant-124,that antagonizes the action of miR-124.Q-RT-PCR analyses demonstrated a significant reduction of endogenous miR-124in cells transfected with the antagomir (Fig.3A).Immunofluorescence analysis revealed that the SOX9protein was absent in the nuclei of XX embryonic gonadal cells when cells were untreated(Fig.3B)or transfected with an unrelated antagomir,Ant-144(Fig.3C).In contrast, SOX9expression was observed in XX gonadal cells transfected with Ant-124(Fig.3D),such that they look similar to control XY gonadal cells(Fig.3E).Q-RT-PCR showed a significant increase in Sox9mRNA in XX gonadal cells transfected with Ant-124but neither in untreated XX cells nor in those transfected with Ant-144(Fig.3F).Overexpression of miR-124Induces Down-Regulation of Sox9in Embryonic Chondrocytes but not in XY Gonadal CellsBased on our hypothesis,overexpression of miR-124would be expected to induce down-regulation of Sox9in cells expressing this gene naturally.To test this possibility,we transfected both cultured chondrocytes and XY gonadal cells with either a precursor miRNA expression vector containing the mouse mmu-mir-124-2gene or a negative control vector containing a scrambled miRNA sequence.The levels of miR-124increased considerably in cells transfected with mmu-mir-124-2,as measured by Q-RT-PCR(Fig.4A).Immunofluores-cence analyses demonstrated that the percentage of SOX9-positive cells was significantly lower in chondrocytes trans-fected with the mmu-mir-124-2expression vector than in those with the control vector.However,this effect was not observed in XY gonadal cells;the percentage of cells expressing SOX9 did not decrease in the presence of the mmu-mir-124-2 expression vector(Fig.4,B–F).Consistent with the results of immunofluorescence studies,Q-RT-PCR analyses showed that overexpression of mmu-mir-124-2reduces the expression levels of Sox9mRNA in chondrocytes,whereas no significant differences were observed in XY gonadal cells(Fig.4G). Because SOX9expression is already robust and maintained by several feedback loops in13.5dpc mouse embryonic testes, miR-124might not be able to efficiently overcome SOX9 expression.Therefore,we repeated the same set of experiments using11.5dpc XY gonadal cells,where Sox9transcription has just started.However,we obtained similar results,confirming the fact that overexpression of miR-124is not sufficient to affect SOX9expression in XY gonadal cells(Supplemental Fig.S3).DISCUSSIONDespite the discovery of the mammalian testis-determining gene SRY some20years ago[1,2],it is thought that some regulatory elements remain unknown,particularly those involved in determining the ovary.Because miRNAs are involved in many developmental processes,it is reasonable to suspect that they may also have some role in gonad development.Several recent studies strongly suggest that this is the case in mammals[35–38].Using miRNA microarray technology,we identified22miRNAs(including mmu-miR-126-5p,mmu-miR-103,and clusters B,C,and D)that are either up-or down-regulated during the critical period of sexual differentiation in a sex-specific manner,indicating that they could participate in gonadal sex differentiation(Fig.1). Interestingly,coincident with ovarian cell determination at 13.5dpc,most of them(16)are up-regulated in the ovary at this stage,suggesting a potential role in ovarian development.Using a clone-based miRNA amplification profiling meth-od,Takada et al.[35]reported that some miRNAs show a sex-dependent expression pattern in13.5dpc mouse gonads. Concerning some of the miRNAs identified by those authors, their results contradict those of our microarray-based screening. Notably,Takada et al.reported that the expression of miR-124 is higher in developing testes than in ovaries[35].However, our data,particularly for miR-124expression,have been repeatedly validated by Q-RT-PCR and in situ hybridization, which is not included in studies of Takada et al.In addition,a recent publication reported that miR-202-5p/3p are differen-tially expressed in the developing testis[43],thus coinciding with our microarray data.In fact,miR-202-3p is included in cluster C and miR-202-5p is in cluster E because the higher expression detected in the testis did not exceed the SD.0.5 threshold(Fig.1).This represents further validation of our microarray results.Several findings support the fact that miR-124could have an important role during ovarian development.First,our miRNA microarray profiling showed that miR-124is differ-entially up-regulated in the XX but not the XY gonad during the transition from11.5to13.5dpc stages.Second,the computational analysis for the miRNAs selected from our microarray results identified potential targets for miR-124in the30-UTR regions of four genes of the male pathway in both human and mouse:Sox8(the SRY-box containing gene8), Sox9,Dmrt1,and AR(androgen receptor).Third,Cheng et al.[42]showed that the mouse miR-124regulates adult neurogenesis by repressing SOX9production in the subven-tricular zone of the brain[42],raising the possibility that this miRNA could act also in the gonad.Overall,these findings strongly suggested that the main role of miR-124in the ovary would be to inhibit the production of SOX9protein.To verify this hypothesis,miR-124was knocked down or overexpressed in primary cultures of embryonic gonadal cells. Transfection with an miR-124-specific antagomir induced the ectopic production of SOX9protein in XX gonadal cells,as well as a significant increase in Sox9mRNA/transcript, implying that miR-124actively represses Sox9in female gonadal cells.The increased expression levels of Sox9in XX cells transfected with antagomir-124are lower than those observed in XY gonadal cells,suggesting that SRY-mediated up-regulation of Sox9is required to reach the male expression level.In the developing mouse testis,Sox9is activated primarily,probably by SF1,in both XY and XX gonads prior to sex differentiation[44].At the time of sex determination, Sox9is up-regulated in the XY gonad by the combined, synergistic action of SRY and SF1on a testis-specific Sox9REAL ET AL.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
BIOLOGIA PLANTARUM 54 (4): 639-646, 2010639Ectopic over-expression of two apple Flowering Locus T homologues, MdFT1 and MdFT2, reduces juvenile phase in ArabidopsisW.M. LI 1,2, Y. TAO 1, Y.X. YAO 1, Y.J. HAO 1 and C.X. YOU 1*State Key Laboratory of Crop Biology and College of Horticulture Science and Engineering, Shandong Agricultural University, Tai-An, Shandong 271018, P.R. China 1South Subtropical Crop Research Institute, Chinese Academy of Tropical Agricultural Science, Zhanjiang 524091, P.R. China 2AbstractTo get insight into mechanism by which apple tree (Malus domestica Borkh .) regulates flowering, two apple flowering locus T (FT ) homologues, MdFT1 and MdFT2, were isolated from the leaf cDNAs of cultivar Gala. The open reading frames (ORFs) of two MdFTs encoded 174 amino acids. The deduced amino acid sequence of MdFT1 and MdFT2 showed 94.3 % similarity to each other, while 72.6 and 76.0 % to AtFT protein, respectively. Semi-quantitative RT-PCR indicated their specific expression in leaves. Visualization of MdFT2-GFP fusion protein demonstrated its localization on membrane. Ectopic overexpression of either MdFT1 or MdFT2 in Arabidopsis significantly induced early flowering by activating the downstream flowering-related genes.Additional key words : apple florigen, early flowering, RT-PCR.IntroductionMany perennial woody plants have a long juvenile phase, spanning from several years to a few decades. In this period, any techniques can not induce to produce flowers or fruits (Hackett 1985, Martín-Trillo and Martínez- Zapater 2002, Poethig 1990). This characteristic may be an advantage for their fully growth and development so that enable them to be competent to adapt various environments. However, in fruit trees, it is a disadvantage severely hampering traditional breeding and genetic studies. In the case of apple (Malus domestica Borkh .), the breeding procedure consists of at least three steps: cross-pollination, seedling selection and regional trials before a variety that meets consumer demands is released. It usually takes 15 or more years. Fortunately, a progress has been achieved during the past decades (Blazquez and Weigel 2000, Colasanti and Sundaresan 2000, Simpson and Dean 2002, Yang et al . 2007).Up to now, our understanding of the genetic control of flowering time mainly comes from studies in Arabidopsis . Several important Arabidopsis regulators that control flowering time have been isolated and characterized. These include FLOWERING LOCUS T (FT ), SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), LEAFY (LFY ), APETALA1 (AP1), FLOWERING LOCUS C (FLC ), TERMINAL FLOWER 1 (TFL1) (Mandel et al . 1992, Weigel et al . 1992, Ohshima et al . 1997, Kobayashi et al . 1999, Michaels and Amasino 1999, Lee et al . 2000). In transgenic Arabidopsis , over-expressing the former four genes caused early flowering (Kobayashi et al . 1999, Lee et al . 2000, Mandel and Yanofsky 1995, Weigel and Nilsson 1995), whereas over-expression of the later two delayed flowering (Ohshima et al . 1997, Ratcliffe 1998). Mean- while, ectopic over-expression of Arabidopsis early- flowering genes in grass and woody plants caused early flowering in some cases, while did not in other cases (Nilsson and Weigel 1997, Rottmann et al . 2000, Penà et al . 2001, González-Schain and Suárez-López 2008). By contrast, ectopic over-expressing the homologues from woody plants usually induced early flowering in Arabidopsis (Kobayashi et al . 1999, Rottmann et al . 2000, Wada et al .2002). Here, a possible explanation is that the⎯⎯⎯⎯Received 18 April 2009, accepted 5 December 2009.Abbreviations : Ct - cycle threshold; GFP - green fluorescent protein; LD - long-day; MS - Murashige and Skoog; ORF - open reading frame; PEPBs - phosphatidylethanolamine binding proteins; SD - short-day.Acknowledgments : This work was supported by Program for New Century Excellent Talents in University (NCET-06-607) and Program for Changjiang Scholars and Innovative Research Team in University (IRT0635). The first two authors contributed equally to this paper.* Corresponding author; fax: (+86) 538 8242364, e-mail: youchunxiang@W.-M. LI et al.640inconsistent effects of these genes might be caused by some specific interactions between the exogenous genes and the host plants (Penà et al . 2001, Martín-Trillo and Martínez-Zapater 2002, Hsu et al . 2006). Hence, for genetic improvement it is necessary to isolate and characterize native genes.Apple is one of the most economically important fruit tree crops in the world. However, little is known in apple tree about genes that control the transition from vegetative to reproductive phase and about the mechanism by which juvenility is modulated. Recently, FT protein, but not FT mRNA, has been proven to be a mobile signal migrated from leaves to the apical meristem to promote floral initiation in several plant species (Yang et al . 2007). MdFT , referring to MdFT1 in this study, was isolated and first appeared in the paper by Kotoda and Wada (2005). More recently, its genomic organization and expression patterns were described by Hättasch et al . (2008). However, it is yet open question if FT homologous genes are involved in flowering regulation in apple tree. To answer this question, two MdFT gene s , MdFT1 and MdFT2, were isolated and identified in this study. Their role in promoting flowering was investigated by ectopic over-expression in Arabidopsis , and then the putative mechanism on its involvement in flowering control was discussed.Materials and methodsApple (Malus domestica Borkh .) leaves, flowers and young fruits of fruit-bearing trees were collected from cultivar Gala in the experimental orchard of Shandong Agricultural University, China. Callus tissues were generated from leaf explants in dark. Arabidopsis thaliana ecotype Columbia (Col) was used in experiments. Seeds were surface sterilized with bleach and sown on Murashige and Skoog (1962; MS) agar medium containing 3 % sucrose. After being stored in dark at 4 °C for 3 d, the seeds were germinated. Seedlings (15-d-old) were then transferred to soil and grown in a growth chamber at ~22 °C under long day (LD; 16-h pho- toperiod) or short day (SD; 8-h photoperiod). Flowering time was measured by scoring the number of rosette leaves of plants grown under SD conditions.Total RNA was extracted from apple tissues and Arabidopsis seedlings using Trizol reagent (Invitrogen Life Technologies , San Diego, USA) according to the manufacturer’s instructions. RNA from each sample (2 μg) was reverse-transcribed by oligo(dT) primer with reverse transcriptase. Gene-specific sense primer MdFT5 (5'-ATGCCTAGGGATAGGGACC-3') and antisense primer MdFT3 (5'-TTATCTTCTCCTTCCACCGG-3'), and sense primer AtFT5 (5'-GATGTCTATAA ATATAAGAGACCCTC-3') and antisense primer AtFT3 (5'-TCTAAAGTCTTCTTCCTCCGCAGC-3') were used to amplify MdFT and AtFT open-reading frames, respectively. The amplified fragments were cloned into pMD18 cloning vector (TaKaRa Biomedicals , Dalian, China) and sequenced for confirmation.Public databases were searched using various Arabidopsis and Oryza sativa FT -related flowering-time genes as queries. Gene names and their GenBank accessions used were as follows: AmCEN (S81193.1) from Antirrhinum majus ; AtATC (NM_128315.2), AtBFT (NM_125597), AtFT (NM_105222), AtMFT (NM_101672), AtTFL1 (NM_120465) and AtTSF (NM_118156.1) from Arabidopsis thaliana ; CmFTL1 (DQ865290) and CmFTL2 (DQ865291) from Cucurbita maxima ; BnTFL1-1 (AB017525) from Brassica napus ; CmoFTL1 (EF462211) and CmoFTL2 (EF462212) from Cucurbita moschata ; CiFT (AB027456) from Citrussinensis ; LeSP (U84140), LeSFT (AY186735.1) and LeSP5G (AY186736) from Lycopersicon esculentum ; MdFT1, MdFT2, MdTFL1-1 (BAD06418) and MdTFL1-2 (BAD10967) from Malus domestica ; NtCET1 (AF145259), NtCET2 (AF145260) and NtCET4 (AF145261) from Nicotiana tabacum ; OsFT (AC136448), OsHd3a (NM_185584), OsMFT1 (NM_183536) and OsRCN2 (XM_472397) from Oryza sativa ; PtFT1 (poplar database number LGVII000284) from Populus trichocarpa ; VvTFL1 (AF378127) from Vitis vinifera ; and TaFT (AY705794) from Triticum aestivum . Multiple alignments of the predicted amino acid sequences were created using software DNASTAR and the resulting alignment was used to generate the phylogenetic tree by the Neighbor-joining method with the bootstrap analysis of 1000 replicates.Semi-quantitative RT-PCR was conducted to analyze MdFTs expression in different apple organs with primers MdFT5 and MdFT3 using the cDNAs of different samples as templates. Ribosomal Md18S RNA was used as loading control. PCR conditions were as follows: 3 min at 94 °C (one cycle), 30 s at 94 °C; 30 s at 53.5 °C and 45 s at 72 °C (30 cycles). The PCR products were run on 1.0 % (m/v) agarose gel and detected using an image analyzer (white/UV transilluminator). To produce 35S::MdFT1, 35S::MdFT2 and 35S::AtFT sense constructs, the cloning vectors were cut with Xba 1 and Sac I to release the MdFT1, MdFT2 or AtFT full-length cDNA fragments. After gel purification, MdFT1, MdFT2 or AtFT genes were ligated in sense orientation into the pBI121 expression vector (TaKaRa Biomedicals ), digested with the same restriction enzymes, downstream of CaMV 35S promoter. The expression constructs were transferred into the Agrobacterium tumefaciens strain LBA3101 with a freeze-thaw method as described by Chen et al . (1994). Transgenic Arabidopsis plants were generated following a floral-dip method according to Clough and Bent (1998). The MdFT2 ORF without stop codon was obtained by RT-PCR using the following primers containing the digestion site (underlined): forward primer 5'-GCT GGATCCATGCCTAGGGATAGGGACC-3'; reverseOVER-EXPRESSION OF MdFT1 AND MdFT2 IN ARABIDOPSIS641primer 5'-GCTGGATCCTCTTCTCCTTCCACCGG-3'. The resulting PCR fragment was digested with Bam HI and cloned into the binary vector pBIN down-stream of the CaMV 35S promoter. The resultant MdFT2-GFP construct was introduced into Agrobacterium strain LBA4404 and transformed into onion epidermal cells which were pre-incubated on MS agar plates in light at 22 °C for 24 h. Infected tissues were incubated on the same conditions for another 24 h, followed by monitoring the subcellular localization of GFP with a confocal laser-scanning microscope (Zeiss LSM 510 META , Jena, Germany).Quantitative real-time PCR was performed in triplicates with SYBR Green PCR master mix (Applied Biosystems , Foster City, USA) using an iQ5 multicolor real-time PCR detection system (BioRad , Hercules, USA). Efficiency of each pair of primers was determined based on its standard curve obtained from a series of 4-fold diluted template cDNAs. The difference between the cycle threshold (Ct) of target genes and the Ct of control primers (ΔCt = Ct target gene - Ct control ) was used to obtain the normalized expression of target genes. Primer sequences used for different flowering time genes were as follows: MdFTs (5'-ACGGATATTCCAGCAACAACTGC-3') and MdFTan (5'-GCGAAACAGCACAAAAACAA-3') for MdFT1 and MdFT2; AtFTs (5'-AACAATCAACACA GAGAAACCACCT-3') and AtFTan (5'-GATCAAG AACGTCTCCAACAACTCT-3') for AtFT ; AtSOC1s(5'-CTCCTCCTATATCTCTACCTATAC-3') and AtSOC1an (5'-GTCACTTGTCTGCTTGTTG-3') for AtSOC1; AtLFYs (5'-GTGGAACCCAACGAGAGCA-3') and AtLFYan (5'-ACCAAGTCGCATCCCAAAAG-3') for AtLFY ; AtAP1s (5'-AGCAGAACCAAGG CCACAA-3') and AtAP1an (5'-TGAGAAAAG GAGATGGCTGATG-3') for AtAP1; AtAGs (5'-TCG CACTCATCGTCTTCTC-3') and AtAGan (5'-GGCTGA TTCTTGTTGATAATACTG-3') for AtAG ; At18Ss (5'-GGAGTTTCAACACATTCTTCGTG-3') and At18San (5'-CCAATCGCCTACCAATACCTTT-3') for At18S.ResultsBlast found that a FT -like full-length cDNA sequence was already registered in GenBank with accession number AB161112 and named as MdFT , however, its function is not characterized yet. To get insight into the role of apple FT -like genes in flowering regulation, RT-PCR was conducted with AB161112 specific primers to amplify the full-length cDNA of MdFT from leaf cDNAs of apple cv. Gala. As a result, a single band was obtained. Following inserting into the cloning vector, six clones were randomly selected for sequencing. Interestingly, those clones fell into two groups depending on their sequences. The sequences were highly similar or exactly same to each other in one group, but relatively different between two groups. Therefore, two different genes existed in the PCR product, and then were designated as MdFT1 and MdFT2, respectively. Their ORFs have the same size and encoded 174 amino acids. Alignment analysis of amino acid sequence showed that MdFT1 was completely identical to AB161112-encoded protein, while MdFT2 shared 94.3 % identity to MdFT1. Both MdFT1 and MdFT2 exhibited high similarity to FT proteins of other plant species (Fig. 1), with 72.6 and 76.0 % identities to AtFT, respectively.As well known, plant FTs belong to CETS protein family which contains conserved amino acid residues or motifs that identical to mammalian phosphatidyl- ethanolamine binding proteins (PEBPs). In MdFT1 and MdFT2, a D-P-D-x-P motif ranges from residue 70 to 74, followed by a histidine residue positioned at 86, and a G-x-H-R motif runs from residue 116 to 119. These motifs are exactly conserved in other members of the CETS family (Fig. 1).Phylogenetic analysis among the deduced amino acid sequences of FT/TFL1-like proteins from different plants showed that MdFT1 and MdFT2 were grouped into the same cluster with other FT-like proteins which shortenjuvenile phase and induce early flowering (Fig. 2). This result suggests that MdFT1 and MdFT2 may potentially accelerate flowering.Due to the high similarity between MdFT1 and MdFT2, it is hard to design specific primers distinguishing one gene from another. Therefore, the total transcripts of MdFT1 and MdFT2 were detected with the primers shared by two genes by semi-quantitative RT-PCR. The result indicated that MdFT transcripts were expressed specifically in leaf, but not in other samples tested such as flower, young fruit and callus (Fig. 3A ).To observe the in vivo localization of MdFT protein, MdFT2, which was more powerful in inducing early flowering in transgenic Arabidopsis than MdFT1, was fused in-frame to the N-terminal side of the green fluorescent protein (GFP) and transiently expressed under control of the CaMV 35S promoter in onion epidermis cells. It was found that GFP fluorescence was presented predominately on membranes, while GFP control constitutively full of the whole cell (Fig. 3B ).To determine the function of MdFTs in flowering regulation, full-length cDNAs of MdFT1 and MdFT2 were inserted in the sense-oriented direction into the plant expression vector pBI121, fusing downstream to the CaMV 35S promoter, then independently introduced into wild-type Arabidopsis plants by Agrobacterium -mediated transformation. In parallel, AtFT was transformed by the same way as a positive control. Under LD conditions, most kanamycin-resistant transgenic seedlings exhibited extremely early flowering even directly flowering in the selection medium and thus failed to produce T1 seeds (data not shown). To harvest seeds, transgenic plants were selected under SD conditions. As a result, more than 10 independent kanamycin-resistant T1 transgenic plants for each 35S::AtFT , 35S::MdFT1 and 35S::MdFT2, respectively, were selected for further investigation.W.-M. LI et al.642Under SD conditions, wild-type Arabidopsis plants flowered with 61 - 73 (average 62.38 ± 3.85 for 8 WT plants) rosette leaves. However, every transgenic line for each gene flowered earlier than the wild type. The earliest flowering appeared in 35S::AtFT transgenic plants bearing only 2 - 5 (3.50 ± 1.09 for 8 transgenic plants) rosette leaves, while the latest in 35::MdFT1 plants with 7 - 12 (8.93 ± 1.54 for 14 transgenic plants) rosette leaves. 35S::MdFT2 transgenic plants flowered early to a medium extent with 3 - 9 (5.41 ± 1.41 for 32 transgenic plants) rosette leaves. The morphology observation also showed that 35S::MdFT2 transgenic plants generally flowered earlier than 35::MdFT1 plants, suggesting that the MdFT2 was more powerful in promoting Arabidopsis floweringthan MdFT1. Therefore, the ectopic overexpression of both MdFT1 and MdFT2 accelerated flowering in Arabidopsis , suggesting their potential function associated with juvenility in apple tree.To get insight into how MdFTs initiate flowering in Arabidopsis , homozygous T2 transgenic lines were selected from the T1 self-cross population for each gene, and then sown for quantitative characterization of flowering time. The transcript levels of four FT -downstream flowering-related genes, i.e. AtSOC1, AtLFY , AtAP1 and AtAG , were examined in response to the ectopic over-expression of AtFT , MdFT1 and MdFT2. The earliest flowering T2 lines for 35S::AtFT and 35::MdFT1, respectively, as well as three 35S::MdFT2 T2Fig. 1. Comparison of amino acid sequences encoded by MdFT1, MdFT2 and other six homologue FT genes from different species. Black boxes indicate identity; crosses and arrowheads stand for D-P-D-x-P and G-x-H-R motif of mammalian PEBPs, respectively.Table 1. Quantitative real-time RT-PCR of transgenes and endogenous genes in WT and transgenic Arabidopsis. The relative expression of AtFT , AtLFY, AtAP1, AtAG and AtSOC1 were relative to those of WT, respectively, and MdFTs to 18S rRNA in 30-d-old seedlings. Plants of each WT, 35S::AtFT and 35S::MdFT1 as well as three 35S::MdFT2 lines with the earliest (3 - 5 rosette leaves), earlier (6 - 7 rosette leaves) and early (8 - 9 rosette leaves) flowering phenotypes, respectively, were grown under SD conditions.Genes WT 35S::AtFT 35S::MdFT1 35S::MdFT2earliest earlier early AtFT1.0 ± 0.3 62.1 ±3.8- - - - AtLFY 1.0 ± 0.1 148.7 ± 24.7 1.4 ± 0.1 17.3 ± 4.1 3.0 ± 0.3 2.6 ± 0.2 AtAP1 1.0 ± 0.3 83.3 ± 4.2 10.8 ± 1.0 17.7 ± 2.3 12.3 ± 1.8 11.8 ± 1.9 AtAG 1.0 ± 0.3 31.4 ± 3.0 23.1 ± 2.1 4.5 ± 0.3 17.0 ± 1.5 14.6 ± 1.6 AtSOC1 1.0 ± 0.4 4.2 ± 0.4 3.3 ± 0.6 1.9 ± 0.3 3.8 ± 1.0 4.7 ± 0.7 MdFTs- - 21.3 ± 9.4 16.9 ± 2.8 8.7 ± 1.8 3.3 ± 0.8OVER-EXPRESSION OF MdFT1 AND MdFT2 IN ARABIDOPSIS643lines with the earliest (3 - 5 rosette leaves), earlier (6 - 7 rosette leaves) and early (8 - 9 rosette leaves) flowering phenotypes, respectively, were chosen for examination with real time RT-PCR. All leaf samples were harvested from 30-d-old seedlings grown under SD conditions before flower buds appeared. The results demonstrated that the transcript abundance of each gene was much higher in transgenic lines than in the wild type (Table 1). Therefore, overexpression of MdFTs accelerated flowering by modulating flowering-related genes in Arabidopsis , suggesting that FT proteins function in a conserved regulation cascade to promoter flowering in different plant species. In addition, it was found among three 35S::MdFT2 transgenic lines that the expression levels of MdFT2 were positively related with the flowering time, i.e . the higher expression, the earlier flowering, further suggesting the function of MdFT2 in promoting flowering.DiscussionMammalian phosphatidylethanolamine binding proteins (PEPBs) bind phospholipids in vitro and inhibit the activity of Raf-1 kinase in vivo (Schoentgen and Jollès 1995, Yeung et al . 1999). These proteins share several regions of notable sequence homology including a D-P-D-x-P motif followed at some distance by a histidine residue and then a G-x-H-R motif, all of which contributeFig. 2. Phylogenetic relationship of FT/TFL1-like protein. The numbers next to the nodes give bootstrap values from 1000 replicates. The genes and their accession numbers are found in Materials and methods.to the conformation of the ligand-binding site (Banfield and Brady 2000). There is also a group of proteins sharing homology to PEBPs in the plant kingdom, which compose CETS family with a suggested function of mediators in a variety of signaling pathways including flowering time control mediated by FT proteins (Pnueli et al . 2001). In this study, it was found that both MdFT1 and MdFT2 contained a D-P-D-x-P motif followed by a histidine residue and a G-x-H-R motif, that are also conserved inother members of the CETS family. Two MdFT genes are highly similar to each other, but 5.7 % difference between them suggests that they should be on different loci in apple genome, maybe arising from its genetic background of high heterozygosity and allopolyploidy (Korban and Chen 1992). A recent study also showed that Populus has two FT-like genes in each species (Hsu et al . 2006). Generally, the presence of multi-alleles is considered as a highly adaptive trait in perennial woody plants because it ensures the reproduction successful in every annual cycle. Our transgenic assay also proved that MdFT1 and MdFT2 offered a non-identical service on the capacity to promote flowering, an observation coincided with other multi-alleles gene in different species (Wada et al . 2002, Lin et al . 2007).The striking advances in genetic control of flowering in Arabidopsis have shown a distinct pathway: CONSTANS (AtCO ), a critical gene that promotes flowering in response to long days (Fowler et al . 1999, Kobayashi et al . 1999, Park et al . 1999), activates AtSOC1 directly and through AtFT promotes flowering (Onouchi et al . 2000, Yoo et al . 2005), whereas AtLFY that directly triggers AtAP1, AtAG and AtAP3 expression acts at least in part downstream of AtSOC1 (Ng and Yanoliky 2000, Yu et al . 2002). According to these clues, we chose AtLFY , AtAP1, AtSOC1 and AtAG as markers to examine whether MdFT1 and MdFT2 positively regulate their expressions. Consistent with the previous studies, our findings showed that ectopic overexpression of either MdFT1 or MdFT2 promoted flowering in Arabidopsis by up-regulating those downstream genes.Since several reports verified that FT protein is mobile as a long-range flowering stimulus (Lifschitz et al . 2006, Corbesier et al . 2007, Jaeger and Wigge 2007, Lin et al . 2007, Mathieu et al . 2007, Tamaki et al . 2007), little doubts have been remained about that FT acts as a florigen. Abe et al . (2005) found that Arabidopsis FT moved from leaves to shoot apex where it interacted with FD and formed FT-FD complex, which in turn triggers AP1 transcription in nucleus, and that FT-EGFP was localized in the nucleus and cytoplasm. In tomato, however, FT ortholog SFT (SINGLE-FLOWER TRUSS) was localized primarily in the nucleus, although weak cytoplasmic staining was detected as well. Meanwhile, unlike Arabidopsis FD, the tomato FD homolog SPGB expressedW.-M. LI et al.644in leaves, making the event of SFT interacting with SPGB happened in leaves. They therefore suggest that SFT triggers downstream systemic signals that regulate growth and flowering (Lifschitz et al . 2006). Therefore, FT orthologs from different plant species are different in subcellular localization and even in the way by which they regulate plant growth and flowering.Fig. 3. Gene expression and protein subcellular localization. A - Semi-quantitative RT-PCR amplification of MdFTs with the cDNA as templates from leaf (LF), flower (FL), fruit (FR) and callus (CA) tissue of apple Gala. Ribosomal Md18S RNA was loading as control.B - Subcellular localization of MdFT2-GFP fusion protein. Transient GFP expression in onion epidermal cells transformed with constructs harboring 35S::MdFT2-GFP or 35S::GFP (as a control). GFP fluorescence was detected constitutively in the whole cell for 35S::GFP construct, but specifically on membrane for 35S::MdFT2-GFP construct (bar = 50 μm).Up to now, the schematic flow of flowering genes in woody plants, especially in perennial woody plants which show great differences in growth habit, shape-size, reproduction behavior with Arabidopsis , is unknown yet. As a perennial woody plant, apple has a long juvenile phase. Although an investigation, which aims at identifying the relationships between the expression of flowering genes and the flower induction and initiation in apple, proves that it has a certain similarity to Arabidopsis (Hättasch et al . 2008), the detailed molecular mechanism by which FT proteins regulate apple flowering remains far from elucidation. In addition, considering the genetic background of high heterozygosity and allopolyploidy (Korban and Chen 1992), it could be hypothesized that apple genome may encode multiple forms of FT proteins to adapt the perennial growth habits and reproduction behavior. In this point, it is an interesting question whether there is apple FT protein with subcellular localization different from its Arabidopsis and tomato counterparts, and how it functions. In this study, our result indicated that MdFT2 protein was predominantly distributed on cytoplastic membrane, distinguishing from the previous findings in herbaceous plants. Further investigation is still needed to elucidate why it is localized in this way and how it regulates the expression of downstream genes.ReferencesAbe, M., Kobayashi, Y., Yamamoto, S., Daimon, Y., Yamaguchi,A., Ikeda, Y., Ichinoki, H., Notaguchi, M., Goto, K., Araki, T.: FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. - Science 309: 1052-1056, 2005.Banfield, M.J., Brady, R.L.: The structure of Antirrhinumcentroradialis (CEN) protein suggests a role as a kinase regulator. - J. mol. Biol. 297: 1159-1170, 2000.Blazquez, M.A., Weigel, D.: Integration of floral inductivesignals in Arabidopsis . - Nature 404: 889-892, 2000.Chen, H., Nelson, R.S., Sherwood, J.L.: Enhanced recovery oftransformants of Agrobacterium tumefaciens after freeze-thaw transformation and drug selection. - Biotechniques 16: 664-670, 1994.Clough, S.J., Bent, A.F.: Floral dip: a simplified method forAgrobacterium -mediated transformation of ArabidopsisOVER-EXPRESSION OF MdFT1 AND MdFT2 IN ARABIDOPSIS645thaliana . - Plant J. 16: 735-743, 1998.Colasanti, J., Sundaresan, V.: ‘Florigen’ enters the molecular age:long-distance signals that cause plants to flower. - Trends Biochem. Sci. 25: 236-240, 2000.Corbesier, L., Vincent, C., Jang, S., Fornara, F., Fan, Q., Searle,I., Giakountis, A., Farrona, S., Gissot, L., Turnbull, C., Coupland, G.: FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis . - Science 316: 1030-1033, 2007.Fowler, S., Lee, K., Onouchi, H., Samach, A., Richardson, K.,Coupland, G., Putterill, J.: GIGANTEA : a circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. - EMBO J. 18: 4679-4688, 1999.González-Schain, N.D., Suárez-López, P.: CONSTANS delaysflowering and affects tuber yield in potato. - Biol. Plant. 52: 251-258, 2008.Hackett, W.P.: Juvenility, maturation, and rejuvenility in woodyplants. - Hort. Rev. 7: 109-155, 1985.Hättasch, C., Flachowsky, H., Kapturska, D., Hanke, M.V.:Isolation of flowering genes and seasonal changes in their transcript levels related to flower induction and initiation in apple (Malus domestica ). - Tree Physiol. 28: 1459-1466, 2008.Hsu, C.Y., Liu, Y.X., Luthe, D.S., Yuceer, C.: Poplar FT2shortens the juvenile phase and promotes seasonal flowering. - Plant Cell 18: 1846-1861, 2006.Jaeger, K.E., Wigge, P.A.: FT protein acts as a long-range signalin Arabidopsis . - Curr. Biol. 17: 1050-1054, 2007.Kobayashi, Y., Kaya, H., Goto, K., Iwabuchi, M., Araki, T.: Apair of related genes with antagonistic roles in mediating flowering signals. - Science 286: 1960-1962, 1999.Korban, S.S., Chen, H.: Apple. - In: Hammerschlag, F.A., Litz,R.E. (ed.): Biotechnology of Perennial Fruit Crops, Biotechnology in Agriculture. Vol. 8. Pp. 203-227. Cambridge University Press, Cambridge 1992.Kotoda, N., Wada, M.: MdTFL1, a TFL1-like gene of apple,retards the transition from the vegetative to reproductive phase in transgenic Arabidopsis . - Plant Sci. 168: 95-104, 2005.Lee, H., Suh, S.S., Park, E., Cho, E., Ahn, J.H., Kim, S.G., Lee,J.S., Kwon, Y.M., Lee, I.: The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis . - Genes Dev. 14: 2366-2376, 2000.Lifschitz, E., Eviatar, T., Rozman, A., Shalit, A., Goldshmidt, A.,Amsellem, Z., Alvarez, J.P., Eshed, Y.: The tomato FT ortholog triggers systemic signals that regulate growth and flowering and substitute for diverse environmental stimuli. - Proc. nat. Acad. Sci. USA 103: 6398-6403, 2006.Lin, M.K., Belanger, H., Lee, Y.J., Varkonyi-Gasic, E., Taoka,K., Miura, E., Xoconostle-Cázares, B., Gendler, K., Jorgensen, R.A., Tony, B.P., Lough, J., Lucas, W.J.: FLOWERING LOCUS T protein may act as the long distance florigenic signal in the cucurbits. - Plant Cell 19: 1488-1506, 2007.Mandel, M.A., Gustafson-Brown, C., Savidge, B., Yanofsky,M.F.: Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. - Nature 360: 273-277, 1992. Mandel, M.A., Yanofsky, M.F.: A gene triggering flowerformation in Arabidopsis . - Nature 377: 522-524, 1995.Martín-Trillo, M., Martínez-Zapater, J.M.: Growing up fast:manipulating the generation time of trees. - Curr. Opin. Biotechnol. 13: 151-155, 2002.Michaels, S.D., Amasino, R.M.: FLOWERING LOCUS Cencodes a novel MADS domain protein that acts as a repressor of flowering. - Plant Cell 11: 949-956. 1999.Mathieu, J., Warthmann, N., Kuttner, F., Schmid, M.: Export ofFT protein from phloem companion cells is sufficient for floral induction in Arabidopsis . - Curr. Biol. 17: 1055-1060, 2007.Murashige, T., Skoog, F.: A revised medium for rapid growthand bioassays with tobacco tissue cultures. - Physiol. Plant. 15: 473-497, 1962.Ng, M., Yanoliky, M.F.: Three ways to learn the ABCs. - Curr.Opin. Plant Biol. 3: 47-52, 2000.Nilsson, O., Weigel, D.: Modulating the time of flowering. - Curr.Opin. Biotechnol. 8: 195-199, 1997.Ohshima, S., Murata, M., Sakamoto, W., Ogura, Y., Motoyoshi,F.: Cloning and molecular analysis of the Arabidopsis gene Terminal Flower 1. - Mol. gen. Genet. 254: 186-194, 1997. Onouchi, H., Igeno, M.I., Perilleux, C., Graves, K., Coupland,G.:Mutagenesis of plants overexpressing CONSTANS demonstrates novel interactions among Arabidopsis flowering-time genes. - Plant Cell 12: 885-900, 2000.Park, D.H., Somers, D.E., Kim, Y.S., Choy, Y.H., Lim, H.K.,Soh, M.S., Kim, H.J., Kay, S.A., Nam, H.G.: Control of circadian rhythms and photoperiodic flowering by the Arabidopsis GIGANTEA gene. - Science 285: 1579-1582, 1999.Penà, L., Martín-Trillo, M., Juárez, J., Pina, J.A., Navarro, L.,Martínez-Zapater, J.M.: Constitutive expression of Arabidopsis LEAFY or APETALA1 genes in citrus reduces their generation time. - Nat. Biotechnol. 19: 263-267, 2001. Pnueli, L., Gutfinger, T., Hareven, D., Ben-Naim, O., Ron, N.,Adir, N., Lifschitz, E.: Tomato SP-interacting proteins define a conserved signaling system that regulates shoot architecture and flowering. - Plant Cell 13: 2687-2702, 2001. Poethig, R.S.: Phase change and the regulation of shootmorphogenesis in plants. - Science 250: 923-930, 1990.Ratcliffe, O.J., Amaya, I., Vincent, C.A., Rothstein, S., Carpenter,R., Coen, E.S., Bradley, D.J.: A common mechanism controls the life cycle and architecture of plants. - Development 125: 1609-1615, 1998.Rottman, W.H., Meilan, R., Sheppard, L.A., Brunner, A.M.,Skinner, J.S., Ma, C., Cheng, S., Jouanin, L., Pilate, G., Strauss, S.H.: Diverse effects of overexpression of LEAFY and PTLF , a poplar (Populus ) homolog of LEAFY /FLORICAULA , in transgenic poplar and Arabidopsis . - Plant J. 22: 235-245, 2000.Schoentgen, F., Jollès, P.: From structure to function: possiblebiological roles of a new widespread protein family binding hydrophobic ligands and displaying a nucleotide binding site. - FEBS Lett. 369: 22-26, 1995.Simpson, G.G., Dean, C.: Arabidopsis , the rosetta stone offlowering time? - Science 296: 285-289, 2002.Tamaki, S., Matsuo, S., Wong, H.L., Yokoi, S., Shimamoto, K.:Hd3a protein is a mobile flowering signal in rice. - Science 316: 1033-1036, 2007.Wada, M., Cao, Q.F., Kotoda, N., Soejima, J.I., Masuda, T.:Apple has two orthologues of FLORICAULA /LEAFY involved in flowering. - Plant mol. Biol. 49: 567-577, 2002. Weigel, D., Alvarez, J., Smyth, D.R., Yanofsky, M.F.,Meyerowitz, E.M.: LEAFY controls floral meristem identity in Arabidopsis . - Cell 69: 843-859, 1992.Weigel, D., Nilsson, O.: A developmental switch sufficient forflower initiation in diverse plants. - Nature 377: 495-500, 1995.Yang, Y.J., Klejnot, J., Yu, X.H., Liu, X.M., Lin, C.T.: Florigen(II): It is a mobile protein. - J. Integr. Plant Biol. 49:。