用MS进行石墨烯建模导入VASP
MS疑难杂症
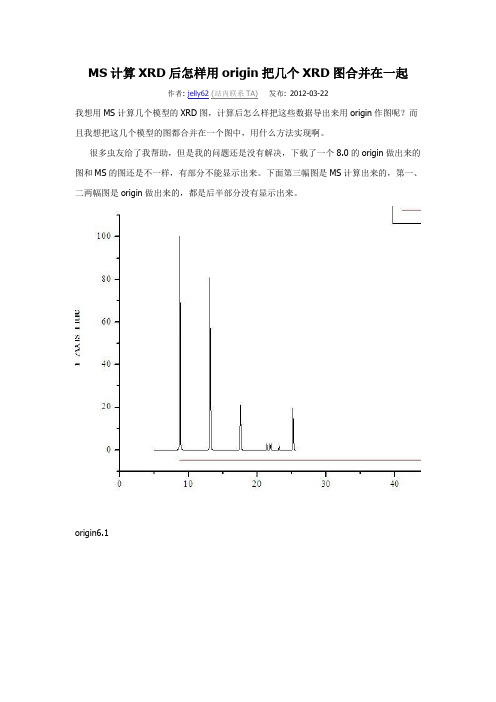
MS计算XRD后怎样用origin把几个XRD图合并在一起作者: jelly62(站内联系TA)发布: 2012-03-22我想用MS计算几个模型的XRD图,计算后怎么样把这些数据导出来用origin作图呢?而且我想把这几个模型的图都合并在一个图中,用什么方法实现啊。
很多虫友给了我帮助,但是我的问题还是没有解决,下载了一个8.0的origin做出来的图和MS的图还是不一样,有部分不能显示出来。
下面第三幅图是MS计算出来的,第一、二两幅图是origin做出来的,都是后半部分没有显示出来。
origin6.1origin8.0MS举报删除此信息贺仪(站内联系TA)你看看这两幅图12楼: Originally posted by 贺仪at 2012-03-22 21:56:26:你是不是没有全选上呀?我确认全选上了,还是这样。
不知道为什么。
贺仪(站内联系TA)13楼: Originally posted by jelly62 at 2012-03-23 09:51:46:我确认全选上了,还是这样。
不知道为什么。
这个可以不选直接复制,你试试看jelly62(站内联系TA)14楼: Originally posted by 贺仪at 2012-03-23 14:28:45:这个可以不选直接复制,你试试看是了,还是一样哦贺仪(站内联系TA)15楼: Originally posted by jelly62 at 2012-03-23 15:22:29:是了,还是一样哦那你用origin是不是启用了speed mode?关闭试试吧……唯美的心(站内联系TA)我也想看看【求助】MS遇到的问题作者: 咖啡喝不醉(站内联系TA)发布: 2010-12-041.discover能量最小化不收敛是什么原因?这样出来的结果可靠吗?2.数据不稳定,是不是体系未趋于平衡呢?怎么解决这个问题呢?举报删除此信息qphll(站内联系TA)如果是有文献可以参照, 那么尽量先采用文献值; 如果是新的体系, 那么很多参数都是需要测试的.你的两个问题其实反映的是同一个本质: 体系还未平衡.建议:(1) 首先做minimization, 使得体系的overlap消除, 这样MD开始的那个初始构型比较合理;(2) 跑足一定的步数, 看能量是否收敛. 这里的收敛并不是要求能量就不变化了, 而是说能量在一个比较小的范围内周期震荡.看你体系大小, 但是一般来讲如果1~2ns以后, 体系能量还是有很大的震荡, 那么需要考虑你的参数的使用是不是合理.其实我也是'空对空', 以后提问记得上图.一图胜千言.咖啡喝不醉(站内联系TA)Originally posted by qphll at 2010-12-05 02:47:49:如果是有文献可以参照, 那么尽量先采用文献值; 如果是新的体系, 那么很多参数都是需要测试的.你的两个问题其实反映的是同一个本质: 体系还未平衡.建议:(1) 首先做minimization, 使得体系的overlap消除 ...此为其中的一个片段,谢谢帮忙再分析一下!043114076(站内联系TA)把结果再优化一下试试咖啡喝不醉(站内联系TA)在做聚乙烯的能量最小化时,聚乙烯的结构是用build直接建成的。
VASP使用总结

VASP计算的理论及实践总结一、赝势的选取二、收敛测试1、VASP测试截断能和K 点2、MS测试三、结构弛豫四、VASP的使用流程(计算性质)1、VASP的四个输入文件的设置2、输出文件的查看及指令3、计算单电能(1) 测试截断能(2) 测试K点4、进行结构优化5、计算弹性常数6、一些常用指令一、赝势的选取VASP赝势库中分为:PP和PAW两种势,PP又分为SP(标准)和USPP(超软)。
交换关联函数分为:LDA(局域密度近似)和GGA(广义梯度近似)。
GGA 又分为PW91和PBE。
在VASP中,其中pot ,pot-gga是属于超软势(使用较少)。
Paw, paw-pbe ,和paw-gga是属于PAW。
采用较多的是PAW-pbe 和PAW-gga。
此外vasp 中的赝势分为几种,包扩标准赝势(没有下标的)、还有硬(harder)赝势(_h)、软(softer)赝势(_s), 所谓的硬(难以赝化),就是指该元素原子的截断动能比较大,假想的势能与实际比较接近,计算得到的结果准确,但比较耗时,难以收敛。
软(容易赝化),表示该元素原子的截断动能比较小,赝势模型比较粗糙,但相对简单,可以使计算很快收敛(比如VASP开发的超软赝势)。
即硬的赝势精度高,但计算耗时。
软的精度低,容易收敛,但节省计算时间。
另一种情况:如Gd_3,这是把f电子放入核内处理,对于Gd来说,f电子恰好半满。
所以把f电子作为价电子处理的赝势还是蛮好的(类似还有Lu,全满)。
(相对其他的4f元素来说,至于把f电子作为芯内处理,是以前对4f元素的通用做法。
计算结果挺好)常用的做法是:用两种赝势测试一下对自己所关心的问题的影响情况。
在影响不大的情况下,选用不含4f电子的赝势(即后缀是3),一来减少计算量,二来避免DFT对4f电子的处理。
【1.赝势的选择:vasp的赝势文件放在目录~/vasp/potentials 下,可以看到该目录又包含五个子目录pot pot_GGA potpaw potpaw_GGA potpaw_PBE ,其中每一个子目录对应一种赝势形式。
用VASP4.6计算晶体硅能带实例

VASP Version : 4.6在此文中,我将用硅晶体作为实例,来说明如何用VASP4.6来计算固体的能带结构。
首先我们要了解晶体硅的结构,它是两个嵌套在一起的FCC布拉菲晶格,相对的位置为 (a/4,a/4,a/4), 其中a=5.4A是大的正方晶格的晶格常数。
在计算中,我们采用FCC的原胞,每个原胞里有两个硅原子。
VASP计算需要以下的四个文件:INCAR(控制参数), KPOINTS(倒空间撒点), POSCAR(原子坐标), POTCAR(赝势文件)为了计算能带结构,我们首先要进行一次自洽计算,得到体系正确的基态电子密度。
然后固定此电荷分布,对于选定的特殊的K点进一步进行非自洽的能带计算。
有了需要的K点的能量本征值,也就得到了我们所需要的能带。
步骤一.—自洽计算产生正确的基态电子密度:以下是用到的各个文件样本:INCAR 文件:SYSTEM = SiStartparameter for this run:NWRITE = 2; LPETIM=F write-flag & timerPREC = medium medium, high lowISTART = 0 job : 0-new 1-cont 2-samecutICHARG = 2 charge: 1-file 2-atom 10-constISPIN = 1 spin polarized calculation?Electronic Relaxation 1NELM = 90; NELMIN= 8; NELMDL= 10 # of ELM stepsEDIFF = 0.1E-03 stopping-criterion for ELMLREAL = .FALSE. real-space projectionIonic relaxationEDIFFG = 0.1E-02 stopping-criterion for IOMNSW = 0 number of steps for IOMIBRION = 2 ionic relax: 0-MD 1-quasi-New 2-CGISIF = 2 stress and relaxationPOTIM = 0.10 time-step for ionic-motionTEIN = 0.0 initial temperatureTEBEG = 0.0; TEEND = 0.0 temperature during runDOS related values:ISMEAR = 0 ; SIGMA = 0.10 broadening in eV -4-tet -1-fermi 0-gausElectronic relaxation 2 (details)Write flagsLWAVE = T write WAVECARLCHARG = T write CHGCARVASP给INCAR文件中的很多参数都设置了默认值,所以如果你对参数不熟悉,可以直接用默认的参数值。
Ag团簇修饰双层石墨烯的弱局域化效应研究的开题报告

Ag团簇修饰双层石墨烯的弱局域化效应研究的开题
报告
一、研究背景:
石墨烯是一种单层碳原子组成的二维材料,由于其优异的电学、热
学等性质而备受关注。
然而,在实际应用中,石墨烯还存在一些问题,
如电子在石墨烯中的弱局域化效应,这需要通过控制石墨烯表面的团簇
来解决。
因此,探究团簇对石墨烯局域化效应的影响,是当前石墨烯研
究的重点之一。
二、研究内容:
本研究将重点探究AG团簇在双层石墨烯上的修饰对石墨烯局域化效应的影响。
通过第一性原理计算,模拟AG团簇在双层石墨烯表面的位置、构型等方面的变化情况,进而分析AG团簇修饰对双层石墨烯电子结构、磁性等性质的影响,并对其机理进行解析。
三、研究意义:
研究团簇修饰对双层石墨烯的弱局域化效应有助于解决石墨烯在应
用中的一些问题,同时也有利于对石墨烯的物理性质进行深入的探究和
理解。
此外,通过本研究的成果,还可以为石墨烯的实际应用提供理论
基础和技术指导。
四、研究方法:
本研究采用第一性原理计算方法,使用VASP软件对双层石墨烯的各种性质进行计算和模拟。
具体包括:构建双层石墨烯和AG团簇的模型,计算石墨烯和AG团簇的结构参数和能量,分析AG团簇修饰对双层石墨烯的电子结构和磁性的影响,并通过谷子和紫外光等实验手段验证计算
结果的可行性。
五、预期成果:
本研究旨在探究AG团簇在双层石墨烯上的修饰对石墨烯局域化效应的影响,并分析其机理。
预计可得到AG团簇修饰对双层石墨烯电子结构和磁性的影响规律,为石墨烯的应用提供理论指导和技术支持,同时也可为石墨烯的物理性质研究提供参考。
MS两种方法构建的石墨烯储氢性能的模拟计算比较

低维碳纳米材料结构性能及应用云南大学2007级物理系物理学专业刘岩学号20071050175石墨烯是一种由碳原子组成的二维六角点阵结构,具有单一原子层或几个原子层厚,具有比较大的比表面积,有做储氢材料的潜质。
本文主要利用Material-studio软件对石墨烯结构和储氢性能进行了一些研究。
Material-studio里有两种构建石墨烯的方法,但是这两种方法构建的原始晶胞却是不同的,而且,相同体积下,结点个数不同,而且直观的看,二者键型有区别。
为了进行对比,本文将两种模型结构和储氢性能分别在相同条件下进行计算和比较。
两种模型的建立方法:第一种,导入软件内置模型执行file – import –structure –ceramics – graphite.msi,获得双层石墨烯,层间距为0.34nm,将其扩充为6层,选定一层,将其移动到模型正中央,模型厚度为0.68*3nm;第二种方法,建立晶胞,选择模型为第183型,设置参数为2.46、2.46和3.4,然后将碳原子添加进去,设置坐标为0.333、0.667和0.500,获得厚度为0.34nm的晶胞,将其扩充为6层,因此它的厚度与第一种一样。
现在要确定两种模型的结点个数,为使体积接近,分别将其扩充为145和128个结点。
如图,显而易见,第一种模型边沿布满结点,而第二种模型边沿没有结点。
为使模型稳定,对它们初步先进行几何结构优化。
优化以前,键角都是120°键长均为0.142nm。
几何结构优化后,键长和键角均发生了一些轻微变化。
(模型一)(模型二)随后,我们对这两种模型设定在77K、10KPa~100MPa进行储氢性能的模拟计算。
这两幅图为石墨烯吸附了氢以后的剖面图,红色点阵为氢可能分布的位置,通过这两幅图,我们可以看到,氢附着于石墨烯时,其分布呈层状,平行于石墨烯,并与之有一定距离。
下图为77K温度下,石墨烯的两种模型对氢吸附能力随压强(10KPa~10MPa)变化的曲线。
《物理建模与仿真实践》课程教学大纲

物理建模与仿真实践
Physical Modelling and Simulation
一、课程基本情况
教学周数:2周
学分:2学分
开课学期:第6学期
课程性质:选修
先修课程:原子物理、计算物理、数学物理方程等
适用专业:物理学、应用物理学
教材:自编讲义
开课单位:物理与光电工程学院物理系
二、实习目标
物理建模与仿真实践是通过计算机把教学内容、教师指导和学生的操作有机的融合为一体,通过对实验环境的模拟,加强学生对物理思想、方法等的理解。
通过本课程的学习,达到培养学生思考能力和比较判断能力,同时也实现了培养动手能力,巩固了之前所学的物理知识,深化了对物理学科认识的目的。
三、实习基本要求
(1)通过本实践课程的学习,让学生掌握物理建模与仿真的基本思想和方法;
(2)通过本实践课程的学习,让学生了解几款本课程常用的软件;
(3)让学生会用VASP等软件对几种常见结构的物质进行建模,并对计算结果进行分析;
四、实习内容及时间安排
五、课程考核
(1)实习报告的撰写要求:每份报告要附结构部分程序
(2)实习报告:7次
(3)考核及成绩评定:平时20%,实习报告80%
六、参考书目
谢希德《固体能带理论》(第二版)复旦大学出版社2007年。
ms中异质结构建和模型计算
ms中异质结构建和模型计算
在MS(Materials Studio)中构建异质结构并进行模型计算,主要涉及以
下步骤:
1. 选择合适的材料:首先,你需要选择两种或多种具有不同导电类型的材料,如P-p结或N-n结等。
2. 确保晶格参数匹配:异质结构建的首要条件是晶格参数的匹配。
如果两种材料的晶格参数不匹配,可能导致异质结变形或垮掉。
因此,要确保晶格参数失配率理论上小于6%。
3. 构建异质结模型:在MS中,你可以直接构建异质结模型。
对于晶格匹配度较高的材料,可以直接构建;对于晶格匹配度较低的情况,需要先找到晶格参数的最小公倍数,然后对两者的晶格参数进行扩胞,再构建异质结。
4. 模型计算:完成异质结模型构建后,可以进行相关的物理性质计算,例如电子结构、光学性质等。
这些计算需要基于量子力学理论,使用合适的计算方法和软件包。
5. 结果分析和优化:计算完成后,需要对结果进行分析和优化。
这包括理解计算结果的意义、比较不同模型的性能、优化模型的参数等。
6. 进一步应用:基于异质结的特性和计算结果,你可以进一步探索其在能源转换、电子器件等领域的应用。
以上步骤是一个基本的流程,实际操作中可能需要根据具体材料和问题进行适当的调整。
同时,建议参考MS的官方文档和教程,以获取更详细和专业的指导。
化学气相沉积生长石墨烯薄膜转移方法及转移用支撑材料的研究进展_蔡伟
过程中采用机械剥离法将 PDMS 与石 墨 烯 分 离,而 石墨烯与氧化硅等 衬 底 的 结 合 并 不 牢 固,所 以 剥 离
过程中石墨 烯 与 PDMS 的 作 用 力 比 石 墨 烯 与 氧 化 PDMS的过程中可能 会 导 致 石 墨 烯 破 损,而 且 该 方
硅衬底的作用力小,从而能使石墨 烯 从 PDMS 上 转 移至氧化硅衬 底 上。2009 年,Hong等 以 [12] 表 面 镀
中,接触面积较小,溶 解 需 要 很 长 时 间,且 溶 解 时 间 PMMA 覆盖在金属基底表面,随后用金属腐蚀溶液
随着石 墨 烯 薄 膜 面 积 的 增 大 而 延 长。 此 外,Hong 腐蚀掉下层的金属基底 得 到 PMMA 与 石 墨 烯 的 结
8
蔡 伟,等:化学气相沉积生长石墨烯薄膜转移方法及转移用支撑材料的研究进展
1.1 溶 解 基 底 法 及 所 用 聚 合 物 材 料
等还提到了一种干 法 转 移,即 直 接 在 镍 基 底 上 覆 盖
在金属基底上 生 长 出 石 墨 烯 薄 膜 后,在 石 墨 烯 一层 PDMS,用 氯 化 铁 溶 液 溶 解 镍 基 底 后 得 到
表面旋涂一层聚合 物 支 撑 层,通 过 腐 蚀 溶 液 将 金 属 PDMS 与 石 墨 烯 的 结 合 体,然 后 进 行 下 一 步 转 移,
CAI Wei 1,2,WANG Cong1,FANG Xiao-hong1,CHEN Xiao-yuan1,YANG Li-you1
(1.Research Center of Thin Film Optoelectronic Engineering,Shanghai Advanced Research Institute, Chinese Academy of Science,Shanghai 201210,China;
VASP单机操作流程(以Ni111表面为例)
八,能带计算(bands):
建立bands文件夹,输入命令:mkdir bands
将KPOINTS INCAR POSCAR POTCAR CHG CHGCAR WAVECAR复制到bands文件夹中,
计算完成后,输入命令:cat Etot.dat查看结果。如同截断能的优化一样,根据要求选择合适的K点。(在此我选择17)
四,优化晶格常数:
在优化晶格常数时,是对比例系数的优化,不能使用绝对值。一般在试验值的左右各取一些值,根据要求选择最合适的晶格常数。计算过程和上两步类似。
Cp En . sh POS . sh(为方便使用格式)
打开终端,编辑K点文件KPOINTS(在此使用自动生成的方法)。
Cat >KPOINTS<<!
>A
>0
>7 7 1
>0 0 0
>!
保存退出。
二,优化截断能:
优化过程的脚本为:
#!/bash/sh
for i in 200 250 300 350 400 450 500 550 600
do
cat >INCAR<<!
(其它内容省略)
(保存退出)
建立relax文件夹:mkdir relax
将CHG CHGCAR WAVECAR INCAR POSCAR KPOINTS POTCAR复制到该文件夹:
Cp CHG CHGCAR WAVECAR INCAR POSCAR KPOINTS POTCAR relax
则在relax文件夹下运行命令;
输入命令:vi INCAR
ms计算模拟石墨烯导热系数的方法
有关“ms计算模拟石墨烯导热系数”的方法
石墨烯的导热系数可以通过多种方法计算,其中包括基于声子传输的理论模型和基于非平衡分子动力学(NEMD)的模拟方法。
有关“ms计算模拟石墨烯导热系数”的方法如下:1.基于声子传输的理论模型:石墨烯依靠声子(晶格振动简正模能量量子)进行热传
输,以弹道—扩散方式传递热量。
其导热系数k可以通过公式k=13Cvl得出,其中C 为声子比热,v为声速,l为平均自由程。
在这个模型中,声子比热、声速、平均自由程这三个参数是关键。
由于石墨烯中碳碳之间的共价键强而碳原子质量小,声子具有较高的声速,因此其导热系数大。
但需要注意的是,声子的比热和平均自由程受温度和尺寸影响较大,声子比热随温度的升高而增大。
2.基于非平衡分子动力学(NEMD)的模拟方法:这是一种更为直接的计算石墨烯导热
系数的方法。
它通过计算物质微小分子在温度变化作用下的运动轨迹和速度,进而得出材料的热传导性能。
采用此方法计算得出的石墨烯垂直导热系数约为
(700±50)W/mK,这一结果表明,石墨烯在导热方面表现出了极高的性能。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
用material studio构建石墨烯模型
弄了好久,终于会构建石墨烯模型了,看了好多帖子,觉得都写的不是很详细,对于新手来说,有些困难,所以特发此贴。
越简单的问题越容易被人忽略~
1、打开material studio,新建一个工程,导入石墨graphite.msi(也可以自己build,然后添加原子)。
2、build->make p1(目的是消除对称性,这样才能够删除一层原子)。
3、删除一层原子(选中原子->delete)。
4、修改晶格参数:build->crystal->rebuild crystal,设置方位角α=90。
,β=90。
,γ=60。
5、构建supercell(方便掺杂,也为了好看):build->symetry->supercell,构建一个5x5x1的超原胞。
6、cleave surface(为了能够添加真空层):build->surface->cleave surface,(h,k,l)改为(0,0,-1)
7、添加20埃真空层(添加真空层是为了减小层与层之间的影响,至少20埃,大点没关系,最多是计算时时间长一点):build->srystal->build vacuum。
构建好后,模型如下:
接下来,在export导出*.p的文件,就可以用于vasp计算了。
最近为了在VASP中弛豫石墨烯,要在在MS中建立石墨烯模型,总结了一下步骤,仅供参考:
1、打开material studio,新建一个工程,import石墨graphite.msi,在structure/ceramic 中
2、build->make p1(目的是消除对称性,这样才能够删除一层原子)。
3、删除一层原子(选中原子->delete),并移动剩下的原子到中间。
4、构建supercell(方便掺杂,也为了好看):build->symetry->supercell,构建一个6*6*1的超原胞,这样建立的超胞模型是扶手型。
要建立锯齿状的,在建立supercell之前,build>symmetry>find symmetry,然后impose s ymmetry,此时只显示两个原子,然后建立supercell,6*6*1
(如果需要supercell是斜六面体,这样就可以了,如果需要矩形,继续下一步)
5、修改晶格参数:build->crystal->rebuild crystal,设置方位角和length。
由斜六面体改为矩形首先改方位角,如果只改方位角,会发现周期性边界变化,
所以还要修改length,修改以后变成矩形。
我的是6*6*1,a:12.78,b:12.2999
6、下面就是需要导出到VASP中
点击castep计算energy,保存file,到相应文件夹下找到graphite (66-cub).cell文件,此文件为隐含文件,打开,把相应的lattice parameter和坐标拷到POSCAR,就搞定了。
一、
1 先导入石墨(陶瓷文件里有),然后build---surface cleave,此时注意hkl的选取以及uv 的方向的选取;然后点击确定即可。
2加真空层的厚度,在BUILD--CRYSTAL 有vacumm选项,注意厚度的选择,一般大于15,3然后做超胞即可,superwell选项
二、
导入graphite (in the file of structure-ceramics ),
然后build -Symmetry-make P1.
然后切掉中间一层。
然后切掉其中一层,将剩下的一层移到c轴中间附近。
bulid-Symmetry-find symmetry-impose symmetry。
这样就得到了honeycomb lattice primitive cell。
如果需要大片结构,可以用bulid - symmetry - Surpercell 来构建。
得到菱形格子。
如果需要正交格子,将primitive cell 通过supercell 2*2 然后,然后留下立方格子的4个原子,其他切掉。
重新构建晶格,build-rebuild 晶格参数,a=4.26,b=2.46 ,c=15。
(通过几何换算得出,或者又构型量出)角度全为90度。
如果需要更大体系,同菱形,用supercell 创建。