常见系统发育软件使用
phylosuite使用介绍

phylosuite使用介绍
PhyloSuite是一个用于分析和编辑分子进化数据的软件套件。
它是由中国科学院遗传与发育生物学研究所的谢威开发的。
该软件套件包括多个工具,可以用于序列比对、系统发育分析、基因结构预测、进化分析等多个方面。
PhyloSuite包含的工具如下:
1. SeqKit:一个用于序列处理的工具,可以进行序列格式转换、过滤、去除冗余等操作。
2. PRANK:一个用于多序列比对的工具,支持DNA和蛋白质序列比对。
3. PhyML:一个用于构建系统发育树的工具,支持多种模型,包括HKY、GTR等。
4. RAxML:一个用于构建系统发育树的工具,支持多种模型,包括GTR、CAT等。
5. Gblocks:一个用于去除进化序列中的低质量区域的工具,可以用于预处理比对序列。
6. Simple_phylogeny:一个用于构建简单系统发育树的工具,可以根据输入序列构建NJ树、UPGMA树等。
7. PhyloFlash:一个用于高通量测序数据分析的工具,可以用于构建系统发育树。
8. OrthoFinder:一个用于寻找同源基因的工具,可以用于基因家族分析。
9. Evolview:一个用于可视化系统发育树的工具,可以用于构建交互式系统发育树。
PhyloSuite提供了一个用户友好的图形界面,用户可以使用鼠标拖拽文件进行操作。
同时,也支持命令行操作,可以更加灵活地控制软件的使用。
PhyloSuite是一个免费的软件,可以在Windows、Mac OS、Linux等多个操作系统上使用。
最新MEGA5使用说明

1MEGA软件——系统发育树构建方法(图文讲解)22012年12月02日⁄Evolution⁄字号小中大⁄评论 3 条⁄阅读 3,872 3次[点击加入在线收藏夹]4一、序列文本的准备5构树之前先将目标基因序列都分别保存为txt文本文件中(或者把所有序列6保存在同一个txt文本中,可以用“>基因名称”作为第一行,然后重起一行编7辑基因序列),序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称8可以已经您的想法随意编辑。
910111213二、序列导入到Mega 5软件14(1)打开Mega 5软件,界面如下151617(2)导入需要构建系统发育树的目的序列1819202122OK23选择分析序列类型(如果是DNA序列,点击DNA,如果是蛋白序列,点击24Protein)252627出现新的对话框,创建新的数据文件282930选择序列类型313233导入序列34353637383940导入序列成功。
41(3)序列比对分析424344点击工具栏中“W”工具,进行比对分析,比对结束后删除两端不能够完全对45齐碱基464748(4)系统发育分析495051关闭窗口,选择保存文件路径,自定义文件名称525354三、系统发育树构建555657根据不同分析目的,选择相应的分析算法,本例子以N—J算法为例585960Bootstrap 选择1000,点击Compute,开始计算616263计算完毕后,生成系统发育树。
646566根据不同目的,导出分析结果,进行简单的修饰,保存67本方法来自网络,经小编microibs编辑,修改补充,如果转载请注明PLoB 68出处。
69。
Mesquite操作步骤
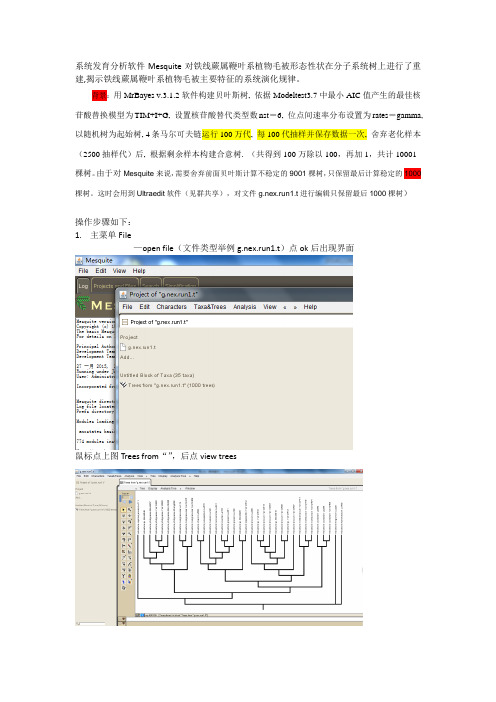
系统发育分析软件Mesquite对铁线蕨属鞭叶系植物毛被形态性状在分子系统树上进行了重建,揭示铁线蕨属鞭叶系植物毛被主要特征的系统演化规律。
背景:用MrBayes v.3.1.2软件构建贝叶斯树, 依据Modeltest3.7中最小AIC值产生的最佳核苷酸替换模型为TIM+I+G, 设置核苷酸替代类型数nst=6, 位点间速率分布设置为rates=gamma, 以随机树为起始树, 4条马尔可夫链运行100万代, 每100代抽样并保存数据一次, 舍弃老化样本(2500抽样代)后, 根据剩余样本构建合意树. (共得到100万除以100,再加1,共计10001棵树。
由于对Mesquite来说,需要舍弃前面贝叶斯计算不稳定的9001棵树,只保留最后计算稳定的1000棵树。
这时会用到Ultraedit软件(见群共享),对文件g.nex.run1.t进行编辑只保留最后1000棵树)操作步骤如下:1.主菜单File—open file(文件类型举例g.nex.run1.t)点ok后出现界面鼠标点上图Trees from“”,后点view trees2.主菜单Characters—New Empty Matix,根据需要填写Number of characters,后点ok而后编辑性状名称,以及更改问号(如铁线蕨毛被:叶轴近轴面用RB表示,叶轴远轴面用RA表示,无毛:0;毛少:1;多毛:2,未知的用?)之后鼠标点击靠左边打开的Tree from“g.nex.run1.t”,出现如下界面3.主菜单Analysis:Tree—Trace characters over trees点ok后,再点ok,,出现如下界面,选择第二个最大释然法Likelihood Ancestral States(相对更可靠),也可以用第一个最大简约法在点击第二个stored probability Model点击Shade states,再点击Stored trees,在如下图中选择Mk1(est.)出现如下界面4.主菜单Display—Tree Form—Balls&Sticks(卫然老师推荐这种树的形状,也可以选择其他的)5.主菜单Trace Over Trees—Show Fraction Of Trees with Equivocal将其勾掉,即不要√6.主菜单Trace Over Trees—Calculate—Average Frequencies Across Trees注意:选择第3步中的最大释然法Likelihood Ancestral States不能识别树的拓扑结构,所以需要手动筛选下图中的Tree #,填写数字范围是1—1000(对应背景),以此来筛选最接近你真实的拓扑结构。
phylosuite使用介绍

phylosuite使用介绍PhyloSuite是一个用于分析系统发育的软件套件,可以在Windows、Linux和Mac OS X 平台上使用,并且支持各种分析方法和文件格式,包括序列比对、物种树、基因树的推断和可视化等功能。
以下是PhyloSuite使用的详细介绍。
1. 下载和安装PhyloSuitePhyloSuite是一个免费的软件,可以从其官方网站下载。
下载完成后,将PhyloSuite 解压缩并打开PhyloSuite的主界面,就可以开始使用PhyloSuite进行系统发育的分析了。
2. 总体流程使用PhyloSuite进行系统发育分析的流程大致分为以下五个步骤,具体如下:(1) 准备数据。
将需要分析的序列数据导入到PhyloSuite中,并将其按照要求进行格式转换和校准等操作。
(2) 序列比对。
使用PhyloSuite内置的多种比对工具进行数据的全局比对、局部比对和进化模型选择等操作,以得到高质量的序列比对结果。
(3) 系统发育分析。
在序列比对的基础上,使用PhyloSuite内置的多种方法推断物种树或基因树,并进行支持率计算和进化树的可视化等操作,以得到最终的系统发育信息。
(4) 结果评估。
对系统发育分析的结果进行统计分析、可视化评估和相关统计检验等操作,以确认分析的准确性和稳定性。
(5) 结果呈现和再利用。
将系统发育分析的结果导出并保存为文本或图片格式,以便用于科学研究、学术会议和出版文章等应用场景。
3. 具体操作在使用PhyloSuite进行系统发育分析时,需要了解各个功能模块的具体操作方法,以下是PhyloSuite中几个核心模块的简要介绍。
(2) Alignment模块。
该模块提供了多种序列比对工具,包括MAFFT, RAxML, MUSCLE 等,并提供多种比对质量评价和修改工具,以得到高质量的序列比对结果。
(3) Phylogeny模块。
此模块支持多种系统发育分析方法和工具,包括Maximum Likelihood, Bayesian Inference, Distance-based等,其可视化输出还包括多种图形化展示方式,以便于直观理解分析结果。
MEGA-系统发育树-快速入门

系统发育树
1.软件准备
DNAman、MEGA
2.序列文件转换格式
2.1先准备一个txt记事本文件,在序列的上一行添加字符>和名称(如>R31-ITS1),然后用MEGA打
开seq格式的序列文件,复制序列到名称下一行
2.2将所需要的比对的所有序列以相同方式写入同一个txt文件中
2.3在MEGA中用ALIGN功能打开准备好的txt文件,选择create a new alignment,数据类型选DNA,
然后从编辑edit中导入新的序列文件(即txt文本)即可导入所需序列
2.4删除无关序列后,先对序列进行分析然后再把序列对齐,类型选DNA,参数默认
颜色一致即为对齐,不一致的就是突变的位点。
然后通常需要把首尾两端没有对齐的序列删掉(只处理首尾两端未对其的序列)
对齐部分
未对齐的删掉
2.5处理完后保存文件并关闭当前窗口,如果不是连续使用的话,切换不同功能时一般点close date
关闭之前的数据
3.构建系统发育树
3.1邻接法构建系统发育树。
生物信息学填空题

填空题:1、蛋白质结构数据来源:①实验测定方法: X-ray 、 NMR 、Cryo-EM ②理论预测:同源建模、折叠识别、从头计算2、一级数据库:①一级核酸数据库:Genbank(美国)、EMBL (欧洲)、DDBJ(日本) NCBI②一级蛋白质序列数据库:SWISS-PORT 、PIR 、 NCBI③一级蛋白质结构数据库:PDB、 pfam 、 prosite大分子序列格式:fasta数据库基本文件格式:genbank蛋白质分类数据库:SCOP、CATH 、 FSSP二次数据库: GDB 、 Prosite、 TRANSFAC3、本地软件: Clustal-x 、 BioEdit 、 Mega、 sequencher、 spdbv、 Discovery-studio4、本课程主要理论依据:相似性、同源性、序列比对(3D结构比对)、数学方法、分子动力、分子力学5、基因鉴定三步骤:①找到序列中的非编码区(低复杂度区)②找基因③鉴定找到的基因6、主要的生物大分子数据:①DNA:基因组序列、基因序列、cDNA、EST、碱基修饰DNA 功能模块 /位点(如启动子、剪接体、表达调控位点等)②蛋白质:氨基酸组成、氨基酸序列、理化性质、原子坐标;二级结构、核体、结构域、功能域 /位点; 3D 结构常见的生物信息数据记录格式:FASTA 、GenBank、EMBL、 PDBFASTA 格式:序列文件的第一行由大于符号>大头的任意文字说明,主要为标记序列用。
从第二行开始是序列本身,标准核苷酸符号或氨基酸单字母符号,通过核苷酸符号大小写均可,而氨基酸一般用大写字母。
文件中和每一行都不要超过80 个字符(通常60 个字符)GenBank格式:序列名称、长度。
日期;序列说明、编号、版本号;物种来源、学名、分类60学位置;相关文献作者、题目、刊物、日期;序列特征表;碱基组成;序列本身(每行个)二 .填空题1.常用的三种序列格式: NBRF/PIR,FASTA 和 GDE2.初级序列数据库: GenBank, EMBL 和 DDBJ3.蛋白质序列数据库: SWISS-PROT 和 TrEMBLPIR (蛋白4. 提供蛋白质功能注释信息的数据库:KEGG (京都基因和基因组百科全书)和质信息资源) 5. 目前由 NCBI 维护的大型文献资源是PubMed6.数据库常用的数据检索工具: Entrez, SRS, DBGET7.常用的序列搜索方法: FASTA 和 BLAST8.高分值局部联配的 BLAST 参数是 HSPs(高分值片段对), E(期望值) 9. 多序列联配的常用软件: Clustal10.蛋白质结构域家族的数据库有:Pfam, SMART11. 系统发育学的研究方法有:表现型分类法,遗传分类法和进化分类法12. 系统发育树的构建方法:距离矩阵法,最大简约法和最大似然法13. 常用系统发育分析软件:PHYLIP 14.检测系统发育树可靠性的技术: bootstrapping 和 Jack-knifing 15. 原核生物和真核生物基因组中的注释所涉及的问题是不同的16. 检测原核生物ORF 的程序: NCBI ORF finder17. 测试基因预测程序正确预测基因的能力的项目是GASP(基因预测评估项目)18.二级结构的三种状态:α螺旋,β折叠和β转角19.用于蛋白质二级结构预测的基本神经网络模型为三层的前馈网络,包括输入层,隐含层和输出层20.通过比较建模预测蛋白质结构的软件有SWISS-PDBVIEWER ( SWISS — MODEL 网站) 21. 蛋白质质谱数据搜索工具:SEQUEST 22. 分子途径最广泛数据库:KEGG23. 聚类分析方法,分为有监督学习方法,无监督学习方法24. 质谱的两个数据库搜索工具:1、 SEQEST 和 Lutkefi 三大数据库:核酸序列数据库、蛋白质序列数据库、结构数据库世界三大核酸序列数据库:GenBank、 EMBL-Bank 、 DDBJ蛋白质序列数据库:Swiss-Prot、 TrEMBL 、UniProt蛋白质结构数据库:PDB 、SCOP、CATH2、 GenBank 文献、提供了提供的服务:提供了EntrezBLAST 序列类似性检索。
几种常用生物分析软件的特点及其使用简介

几种常用生物分析软件的特点及其使用简介韦荣编1 邱高峰1 张源2(1上海水产大学渔业学院,上海200090;2上海水产大学工程学院,上海200090)摘 要: 基于代表性、实用性的原则选择了RAPDistance ,PHY L IP ,MEG A ,TREECON ,AMOVA ,DIPLO 2MO 和MAPMA KER 等7种系统发育及遗传图谱构建方面的免费共享生物分析软件,简要介绍了它们各自的特点、功能、使用及获得的方法。
以期这些软件对于从事生物技术研究的人员具有一定的指导性,可操作性。
关键词: 软件 生物信息学 使用简介Features and B rief Introduction to the Applicationof Some Common Used Biosoft w aresWei Rongbian 1 Qiu G aofeng 1 Zhang Yuan 2(1Fisheries College ,S hanghai Fisheries U niversity ,S hanghai 200090;2Engi neeri ng College ,S hanghai Fisheries U niversity ,S hanghai 200090)Abstract : In this article ,some common used free biosoftwares about phylogeny and genetic map constructing ,i.e.RAPDistance ,PHY L IP ,MEG A ,TREECON ,AMOVA ,DIPLOMO and MAPMA KER are selected based on the prin 2ciple of representativity and utility to briefly introduce their features ,function ,application and means of getting them.The methods in the article are expected to be instructive and operational for the researchers on biotechnology.K ey words : S oftware Bioinformatics Application introduction 随着IT 技术的不断发展,它已经越来越深刻地渗透到各门学科中,包括生命科学。
phylosuite使用介绍

phylosuite使用介绍
PhyloSuite是一款功能强大的多功能序列分析软件,它能够处
理序列数据、进行系统发育分析以及执行其他相关的生物信息学任务。
PhyloSuite包含多个模块,包括序列预处理、进化树构建、比对、
进化树可视化、SNP分析等。
使用PhyloSuite进行序列预处理时,用户可以将多个fasta文
件合并成一个大文件,进行序列长度筛选、去重、筛选保守区域等预处理操作。
进化树构建模块支持多种方法,包括最大似然法、贝叶斯法、邻接法等,并可以选择模型和分支支持率阈值。
比对模块支持多种比对算法和参数设置,例如MAFFT、MUSCLE、CLUSTAL等,还可以
进行多序列比对和序列修剪。
进化树可视化模块可以根据不同的需求生成不同类型的进化树图形,例如圆形树、方形树、分支颜色标记等。
PhyloSuite还提供SNP分析模块,可以对序列中的SNP检测、
过滤、注释等。
此外,还可以进行基因注释、基因组注释、基因组结构预测等其他功能。
使用PhyloSuite,用户可以快速、准确地进行
序列分析和系统发育分析,为生物学研究提供有力的支持。
- 1 -。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
常见系统发育软件使用方法Xie Lei BJFU1 Paup MP流程: Mac准备nex文件(interleave和noninterleave均可) → 存入新建文件夹→拖入paup或用paup打开→ execute → log file → cstatus → tstatus → hsearch → define outgroup → roottrees → savetrees → describetrees →contree(save to file) →save pict→bootstrap(save tree file) →print bootstrap tree→save pict. →stop log.PC版操作,可将附录批处理文件容粘贴至nex文件后面,execute即可。
2 Paup ML 流程:Mac准备nex文件(interleave和noninterleave均可) → 存入新建文件夹→拖入paup或用paup打开→execute→从modeltest软件中打开paupblock运算检测模型→生成score file→打开modeltest中的bin读取score数据→生成结果文档→存档并打开此文档→AIC→将begin paup的运算模块贴至原nex数据文件后面→重新将其拖入paup运行→选择ML运算模式→hsearch→打印树图→save pict. →bootstrap.PC版操作,可将附录5批处理文件容粘贴至nex文件后面,execute即可。
3 Garli运算ML流程:准备nex文件(interleave) → 存入新建文件夹→拖入paup或用paup打开→execute→输出noninterleave文档(若直接是noninterleave上述过程省略,又如果是PC机paup,无菜单操作,可在paup命令行中输入附录1*的命令回车即可生成noninterleave数据)。
使用noninterleave文档(数据中类群名称不得有单引号,空格,所有方括号中容删除)→新建文件夹存入→按照流程2进行modeltest→在苹果机上打开Garli→导入数据→把model定好→run(切记此处不要激bootstrap选项)将上次运算数据拷贝至一新建文件夹→导入苹果版Garli→激活bootstrap选项→定好model→run所有结果用paup软件打开→save pict→打开bootstrap树→做50% majority rule contree→save pict.注:Garli苹果和PC版都有,但是操作不同。
数据格式:和算PAUP一样的nexus格式,但是这个格式有很多注意事项,一些常见的小错误会造成软件无法运行。
参见下列常见问题:1 一定要noninterleave的数据,否则软件无法运算2 [ ]虽然在mrbayes和paup中不成问题但是在garli中有影响,里面容在算之前全部删除为好。
3 taxon名称中可以有下划线但是不得有空格,逗号句点等,否则无法运行。
Mac版GUI的菜单界面,只要有上述正确的nexus格式的数据文件即可运算。
PC版Nex format plus a command file每次使用时拷贝一个软件的文件夹,将此文件夹重新命名(尽量清楚易查询)。
将正确的数据文件拷贝到此文件夹下(与garli运行程序在同一目录下)。
编辑命令文档(名称是garli),进行参数设置。
完成后双击garli运行程序图标即可运算。
4 Bayes 流程Noninterleave 文件→贴运算程序到文件后面(见附录)→将其拷贝至MrBayes 文件夹下→打开运行程序→execute 文件名.扩展名→运算结束后用paup运行源文件→从.t文件中取树→burnin→做50% majority rule contree→save pict.5 r8s流程按照流程2进行modeltest→按照流程2进行ML运算→运算结束print tree→检查这棵带枝长的树是否有分支长度为0的分支→如果有在restore和delete taxa中将这些类群去除→存储带枝长的树到file(nex格式) →将树的taxa名称更换为实际类群名称→将树文件贴至运算模版(见附录)→首先进行第一步cross validate→得到smooth值→替换smooth值再算一遍即可。
注:r8s:该软件与BEAST不同,是对已存在的树进行操作,校订分子钟。
算法为PL法。
先选择模型算出一个ML树(要带枝长),注意如果要用这个树算r8s要保证该树各个分支清晰,有较高的分辨率,没有0枝长分枝和polytomy。
在这个树的tre的nexus文件上面编辑各种命令,然后输入r8s进行运算。
一定要固定root,ingroup最好有一个以上的constraints.运算两次,第一次为cross-validation得出smooth value,第二次再设置这个值算出最终结果。
6 BEAST流程Noninterleave 文档→ BEAUTI打开→ 定义节点→基本设置→化石点标定→生成xml文档→BEAST打开运算→运算完成→Tacer打开log文件→TreeAnnotator 打开树文件→生成out文件→Figtree打开out文件。
7 DIV A在数据文件中确定树形,标注分布区,然后运算1 open DIV A2 proc filename.txt;3 optimize;Batch: optimize maxareas=8 weight=0.5 bound=200 hold=10000;8 Haplotype network简明流程:1、NETWORK 4.5用DNASP软件打开fas或者nex文件→ 另存为Roehl File Format文件(rdf格式)→ 用NETWORK打开→ 进行编辑(更改名称、连续的gap合并、添加删除序列等)后保存→ 选择Median Joining进行calculate network → 在新出现的窗口中导入rdf文件(也可以在这一步直接导入noninterleave的nex文件而省去以上步骤,但是不能重新编辑)→ 设置参数后进行计算,生成out格式的输出文件→ 选择draw network,在新窗口中导入out文件→ 按照提示就可以生成network图,保存fdi文件或可另存为bmp或者pdf文件。
2、TCS 1.21准备noninterleave的nex或者phy文件→ 导入TCS → 设置Parsimony Limit 或Fix connection Limit,并定义gap后点击run → 运行完成后就会自动生成network图,编辑后保存GML文件或可另存为postscript 或者PICT文件。
9.推荐使用:Mega 5.全能系统发育分析软件,从序列校对到系统发育分析、分子钟、性状演化、居群分析全能做。
Mesquite 2.7也很全能,但是主要还是做性状演化分析。
McClade是其收费版。
TNT是原来的Hennig86,做系统发育分析。
PAST十分强大的统计软件,居群分析,分类学的morphometric分析很好。
S-DIV A川大开发的带统计功能的地理分析软件。
建议访问:/phylip/software.html附录1.最大简约法分析批处理文件1.begin PAUP;log file=hsearch1.log;set autoclose=yes;set maxtrees=100 increase=auto;hsearch start=stepwise addseq=random nreps=1000 savereps=yes randomize=addseqrstatus=yes hold=1 swap=tbr multrees=yes nchuck=200 chuckscore=1;savetrees file=hsearch1.all.tre brlens=yes;filter best=yes permdel=yes;savetrees file=hsearch1.best.tre;gettrees file=hsearch1.best.tre;contree all/majrule=yes treefile=contree.tre;log stop;end;2.begin PAUP;log file=hsearch1.log;set autoclose=yes;set maxtrees=100 increase=auto;hsearch start=stepwise addseq=random nreps=1000 savereps=yes randomize=addseq rstatus=yes hold=1 swap=tbr multrees=yes;savetrees file=hsearch1.all.tre brlens=yes;filter best=yes permdel=yes;savetrees file=hsearch1.best.tre;gettrees file=hsearch1.best.tre;contree all/majrule=yes treefile=contree.tre;log stop;end;2的策略较好,是hsearch中首选,而如果此程序运算时间太长则用上面一个程序。
本实用方法只提供hsearch,而branch and bound和exhaustive方法省略。
附录1*export format=nexus interleaved=no file=temp.txt (此为生成noninterleave文件命令)附录 2. ILD分析;endblock;charpartition dna=ITS:1-848,trnLF:849-3051;begin paup;set criterion=parsimony;log file=iscap.hom;hompart part=dna nreps=100/addseq=random;hsearch swap=tbr;endblock;附录 3. Bayes分析1. 单一模型begin mrbayes;lset nst=6 rates=gamma;mcmcp ngen=2000000 printfreq=1000 samplefreq=100 nchains=4 savebrlens=yes filename=P_combined;mcmc;sumt filename=P_combined.t burnin=2000;end;Begin mrbayes;2. 多模型begin mrbayes;charset its=1-622;charset trnlf=623-1696;charset matk=1697-3221;charset chs=3222-4406;charset psbm2trnd=4407-5506;charset psbm=5507-6279;charset gpd=6280-6972;charset LFY=6973-8426;partition Names = 8: its, trnlf, matk, chs, psbm2trnd, psbm, gpd, LFY;end;begin mrbayes;[The following lines set up a model in which all four genes have their unique GTR + gamma+ propinv model]set partition=Names;prset ratepr=variable;lset applyto=(1,2,3,4,5,6,7) nst=6;lset applyto=(8) nst=2 rates=gamma;unlink shape=(all) pinvar=(all);end;begin mrbayes;mcmcp ngen=100000 printfreq=1000 samplefreq=100 nchains=4 savebrlens=yes filename=solms8mys;mcmc;sumt filename=solms8.t burnin=3000;end;3. 带支长contree的运算batchbegin mrbayes;log start filename=cp.log;mcmc filename=cp ngen=2000000 samplefreq=100;sump burnin=5000 filename=cp printtofile=yes outputname=cp.sump;sumt burnin=5000 filename=cp contype=allcompat;log stop;end;附录4. bootstrap分析begin paup;log file=bootstrap.log;set maxtrees=100 increase=auto;set criterion=parsimony;set root=outgroup;outgroup 56 57;bootstrap nreps=1000 conlevel=50 treefile=bootstrap.tre keepall=yes cutoffpct=50/start=stepwise addseq=random nreps=100 savereps=yes nchuck=20 chuckscore=5 dstatus=none;log stop;end;附录 5. Paup运行ML分析苹果机:BEGIN PAUP;Lset Base=(0.3271 0.1847 0.1967) Nst=6 Rmat=(0.9731 1.0473 0.2212 0.4660 1.5241) Rates=gamma Shape=0.8160 Pinvar=0.6410;END;PC机:Begin Paup;Set criterion=likelihood;Lset Base=(0.3413 0.2847 0.0895) Nst=6 Rmat=(0.3885 3.5246 0.5305 0.43643.2970) Rates=gamma Shape=0.5232 Pinvar=0.2613; Hsearch start=nj nchuck=2 chuckscore=5 dstatus=none; savetrees format=nexus brlens=yes append=yes file=likelihood; lscores 1/scorefile=likelihood.sf append=yes;set root=outgroup;outgroup 27 28;showtrees all;end;。