96孔板快速准备DNA
磁珠法(多糖多酚)植物组织基因组DNA提取试剂盒
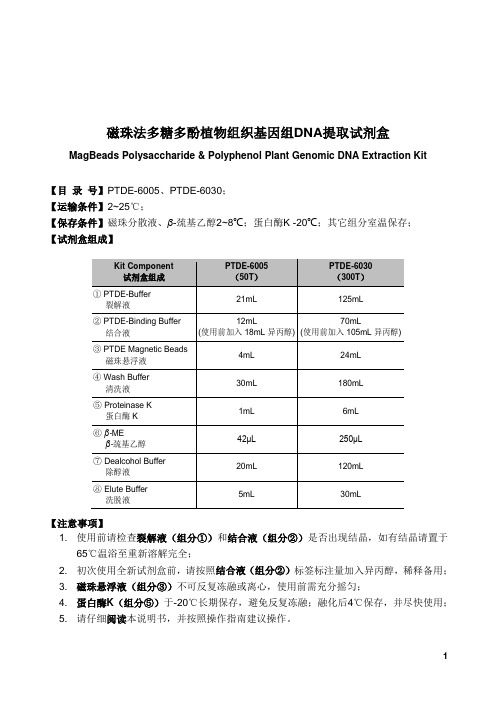
磁珠法多糖多酚植物组织基因组DNA提取试剂盒MagBeads Polysaccharide & Polyphenol Plant Genomic DNA Extraction Kit【目录号】PTDE-6005、PTDE-6030;【运输条件】2~25℃;【保存条件】磁珠分散液、β-巯基乙醇2~8℃;蛋白酶K -20℃;其它组分室温保存;【试剂盒组成】【注意事项】1. 使用前请检查裂解液(组分①)和结合液(组分②)是否出现结晶,如有结晶请置于65℃温浴至重新溶解完全;2. 初次使用全新试剂盒前,请按照结合液(组分②)标签标注量加入异丙醇,稀释备用;3. 磁珠悬浮液(组分③)不可反复冻融或离心,使用前需充分摇匀;4. 蛋白酶K(组分⑤)于-20℃长期保存,避免反复冻融;融化后4℃保存,并尽快使用;5. 请仔细阅读本说明书,并按照操作指南建议操作。
磁珠法·自动化:为生命科学提供自动化磁纳米捕获方案【产品简介】本试剂盒采用针对富含多糖多酚植物组织进行杂质去除的特殊磁珠,配合高性能缓冲液体系,可从各种富含多糖多酚植物组织样本中高质量的分离纯化基因组DNA。
特殊技术包埋的磁珠在特定条件下对核酸具有极强的亲和力,而当条件改变时,磁珠会释放所吸附的核酸,从而达到快速分离纯化核酸的目的。
提取所得的基因组DNA产物片段大、纯度高、质量稳定可靠,尤其适合高通量仪器自动化提取,特别是本公司生产的各类型号自动化核酸提取仪或工作站。
使用本试剂盒纯化所得核酸产物可适用于各种常规分子生物学下游实验,如:酶切、PCR、荧光定量PCR、文库构建、Southern杂交、芯片检测和高通量测序等。
【试剂盒说明】【自备仪器及耗材】研钵&研磨棒(或者研磨机、匀浆机)、水浴锅、涡旋混合仪、高速离心机、EP管(1.5mL 或2.0mL)、EP管配套用磁力架、核酸提取仪(仪器自动版操作步骤需准备)。
【自备试剂】液氮、乙醇(80%, v/v)、异丙醇、RNase A溶液(100mg/mL,分散液10mM Tris-HCl, 1mM EDTA, pH值8.0)。
快速食用菌基因组提取方法

草菇基因组DNA提取方法1.收集菌丝(可提前几天收集)。
用滤纸吸干菌丝,包裹于做好标记的锡箔纸中,迅速扔进液氮中,等所有菌丝收集完毕后,用镊子将样品从液氮中取出,保存于-80℃冰箱备用。
2.称取0.1-0.2g滤纸吸干、包于锡箔纸、冷冻保存的菌丝。
用液氮研磨,在粉末融化前将粉末快速转入用液氮预冷的1.5mL离心管中,快速盖上离心管盖子,扔进液氮中备用(继续研磨下一个样品,快速装进离心管后迅速扔进液氮保存)。
3.用镊子将离心管从液氮中夹出,放在96孔板,快速往离心管中加入400ul提取缓冲液(SDEB),立即涡旋混匀直到变成匀浆(一个一个加样,避免加样前,离心管中的粉末已经融化,涡旋震荡加用手弹使粉末充分溶于SDBE)。
(此时开启离心机,4℃预冷)4.往离心管中加入400ul 2XcTAB缓冲液(2XcTAB)。
5.在通风厨中,往离心管中加入250ul 苯酚,250ul 氯仿:异戊醇(24:1),室温涡旋5分钟。
6.13000 rpm,4ºC,离心5分钟。
(此时可以收拾清洗研钵、收拾擦干桌面)7.小心吸取650ul 上清,转移到一个干净的1.5毫升管中(千万不要吸到任何苯酚或分层处的东西!)。
8.往上清液中加入455ul(0.7倍体积)的异丙醇,上下颠倒混匀10次。
9.13000 rpm,4ºC,离心10分钟。
(此时可以继续收拾清洗研钵、收拾擦干桌面)10.用手倒掉上清,13000rpm,4℃,离心10秒,用200ul 的黄色枪头将离心管中残余的上清弃除。
11.往离心管沉淀中加入900 ul 70%乙醇,用手指弹一弹使白色沉淀悬浮(备选:可放置-20℃冰箱放置1-2h,休息一下,或者去吃饭)。
12.13000 rpm,4ºC,离心10分钟。
(此时可以打开55℃水浴锅)13.用1mL蓝色枪头轻轻吸走上清,注意不要把白色沉淀吸走了(千万不能用手把上清倒掉,一定要用枪轻轻吸)。
质粒dna的提取及电泳检测实验报告

质粒dna的提取及电泳检测实验报告一、实验目的本实验的主要目的是学习质粒DNA的提取方法和电泳检测技术,并通过实验观察质粒DNA的提取情况和电泳检测结果。
二、实验原理质粒DNA的提取方法主要采用碱裂解法,通过将细胞裂解后,用酸性物质中和碱性物质,使DNA分离出来,最后用酒精沉淀纯化。
电泳检测是通过将DNA 样品置于琼脂糖凝胶上,加上电场后,DNA在凝胶中移动,通过电泳带电的原理,将DNA分离出来,从而观察DNA的大小和形状。
三、实验步骤1.准备样品:将含有目标DNA的菌落挑出放入EP管中,加入100μl TE缓冲液(10mmol/L Tris-HCl,1.0mmol/L EDTA,pH8.0),用微量移液器均匀混合。
2.制备裂解液:取10μl 0.25mol/L NaOH加入菌液中,轻轻摇匀,并立即加入200μl 1%SDS(十二烷基硫酸钠)液,翻转混匀。
3.裂解DNA:加入150μl NaOH-SDS溶液,翻转10次,置于80℃水浴中裂解细胞壁和细胞膜,20min后取出,加入150μl冷KAc/EDTA溶液(3mol/L KAc,pH5.2,0.5mol/L EDTA),翻转混匀,冰上静置5min。
4.离心沉淀:10000r/min离心10min,将上清液移至新的96孔板中,加入0.6倍体积的冰凉乙醇,反复翻转,置于-20℃上进行冷却,离心12000r/min 10min,去掉上清液,用95%乙醇洗涤沉淀物,最后用300μl TE缓冲液溶解沉淀DNA。
5.电泳检测:取相同浓度的DNA样品,加入琼脂糖,混合后加入电泳槽中,通电检测10min。
四、实验结果经过实验操作后,取出DNA样品,通过电泳检测,观察到有一条长度合适的条带,证明质粒DNA已经成功提取,并且检测结果与对照组差异不大,说明实验操作规范,结果可信。
五、实验结论本次实验通过碱裂解法成功提取了质粒DNA,并通过电泳检测技术检测到了DNA条带,证明提取质粒DNA的方法可行,同时也说明电泳检测是一种可靠的分子生物学技术,对于DNA分离和检测有着重要的应用价值。
DNA测序操作步骤

D N A测序操作步骤B i g D y e T e r m i n a t o r k i t v3.01.DNA模板的纯度与用量要求DNA纯度:OD260 / OD280 = 1.6 ~ 2.0。
DNA浓度:质粒200 ng / μL,PCR产物10 ng / μL。
DNA用量:PCR产物100-200 bp 1-3 ng200-500 bp 3-10 ng500-1000 bp 5-20 ng1000-2000 bp 10-40 ng> 2000 bp 40-100 ng单链DNA 50-100 ng质粒,双链DNA 100-500 ngCosmid, BAC 0.5-1 μg细菌基因组DNA 2-3 μg2. 测序反应以下加样量以质粒测序为例。
测序PCR产物请按上面表格调整DNA用量,其余条件不变。
Cosmid、BAC和细菌基因组DNA等大分子测序需使用40μL反应体系,所有成分用量加倍。
详细说明请参见BigDye英文手册。
所有试剂从冰箱中取出融化后,稍稍振荡,离心,然后使用。
PCR反应的操作在冰上进行。
试剂用量DNA (200 ng/μL) 1 μLBigDye Mix 8 μL引物 (3.2 pmol / μL) 1 μL灭菌去离子水10 μL总体积20 μL测序PCR热循环条件:(96 ︒C 10 sec → 50 ︒C 5 sec → 60 ︒C 4 min) x 25个循环→ 4 ︒C保温3. 测序产物纯化——96孔板法1. 每管加入80 μL NaAc/酒精混合液,室温放置15 min,最高速(2000-3000 x g)离心30 min(离心机需要配置96孔板转头);马上倒置96孔板,50 x g离心1 min;2. 加入150 μL 70% 酒精,最高速(2000-3000 x g)离心15 min;倒置96孔板,50 x g离心1 min;3. 重复第2步1次。
4. 让残余的酒精在室温挥发干,加入10 μL Hi-Di Formamide溶解DNA。
镍离子亲和层析柱 96孔板

镍离子亲和层析柱 96孔板镍离子亲和层析柱是一种常用的柱层析技术,用于分离和纯化含有镍离子亲和剂的样品。
本文将介绍镍离子亲和层析柱96孔板的原理、应用和优势。
一、镍离子亲和层析柱96孔板的原理镍离子亲和层析柱的原理是基于镍离子与带有特定亲和基团的化合物之间的相互作用。
在柱中填充了含有镍离子亲和剂的固定相,当样品溶液通过柱时,镍离子与亲和剂之间发生络合作用,使目标物质与其他组分分离。
二、镍离子亲和层析柱96孔板的应用镍离子亲和层析柱96孔板广泛应用于生物制药、生物化学、基因工程、蛋白质纯化等领域。
其中一些常见的应用包括:1. 蛋白质纯化:镍离子亲和层析柱可用于从复杂的混合物中高效地纯化目标蛋白质。
通过调节溶液的pH值和离子强度,可以实现目标蛋白质与非特异性结合物质的分离。
2. 重组蛋白表达和纯化:镍离子亲和层析柱可用于表达和纯化重组蛋白。
通过在融合蛋白的表达载体中引入镍离子亲和标签,可以方便地将目标蛋白质与其他细胞组分分离。
3. DNA纯化:镍离子亲和层析柱可用于从DNA样品中纯化目标DNA。
通过使DNA与镍离子亲和剂络合,可以将目标DNA与杂质分离。
4. 药物筛选:镍离子亲和层析柱可用于药物筛选和药物分离。
通过将药物与镍离子亲和剂进行竞争结合,可以评估药物与亲和剂之间的亲和力,从而筛选出具有潜在药物活性的化合物。
三、镍离子亲和层析柱96孔板的优势镍离子亲和层析柱96孔板具有以下优势:1. 高效分离:镍离子亲和层析柱能够高效地分离目标物质,从而提高纯化效率和产量。
2. 灵活性:镍离子亲和层析柱适用于多种样品类型和实验条件。
通过调节溶液的pH值、离子强度和其他条件,可以实现对不同目标物质的高效分离。
3. 经济性:镍离子亲和层析柱具有较低的成本,是一种经济实用的分离纯化方法。
4. 可重复使用:镍离子亲和层析柱可以重复使用,减少实验成本。
5. 适用于高通量筛选:镍离子亲和层析柱96孔板可以与自动化设备配合使用,适用于高通量筛选和生产。
96孔板间接免疫荧光实验步骤

96孔板间接免疫荧光实验步骤96孔板间接免疫荧光实验是一种常用的实验方法,用于检测特定抗原和抗体的结合情况。
以下是该实验的详细步骤,帮助您完成实验并获得准确的结果。
第一步:准备实验材料和试剂在进行实验前,确保准备好所需的试剂和材料,包括:1. 96孔板:用于进行实验的常用多孔板。
2. 抗原:待测物质,可以是细胞表面抗原、蛋白质、细菌等。
3. 抗体:具有高亲合力和特异性,用于与抗原结合。
4. 荧光标记的二抗:可以与抗体结合并发出荧光信号。
5. 缓冲液:用于稀释抗原和抗体的缓冲液。
第二步:包被抗原1. 取一小部分被测抗原,在适当的浓度下进行稀释。
2. 取出96孔板,并将稀释好的抗原加入孔中。
3. 在孔中加入适量的缓冲液,并在室温下静置一段时间,使抗原与孔壁结合。
第三步:阻断非特异性结合1. 弃去孔中的缓冲液。
2. 加入适量的蛋白质(如牛血清蛋白、羊血清蛋白等)作为阻断剂。
3. 在室温下静置一段时间,以防止非特异性结合。
第四步:加入抗体和二抗1. 弃去阻断剂,并用洗涤缓冲液洗涤孔中的抗原。
2. 加入适量的特异性抗体,并在室温下孵育一段时间,使其与抗原结合。
3. 弃去孔中多余的抗体,并用洗涤缓冲液洗涤孔中的抗原。
4. 加入适量的荧光标记的二抗,并在室温下孵育一段时间,使其与抗体结合。
第五步:荧光信号检测1. 弃去孔中多余的荧光标记的二抗,并用洗涤缓冲液洗涤孔中的抗原。
2. 加入适量的检测缓冲液以稀释孔中的荧光信号。
3. 使用荧光读板仪检测每个孔中的荧光强度。
4. 根据结果判断抗原和抗体的结合情况。
第六步:数据分析和结果解释对读取的荧光信号进行数据分析,比较不同孔中的荧光强度。
如果目标抗原与特异性抗体结合,荧光信号应相对较高。
通过对比各孔的荧光强度,可以得出抗原和抗体的结合程度以及目标物质的相对含量。
通过按照以上步骤进行实验,您可以获得准确的间接免疫荧光实验结果。
这种实验方法广泛应用于生物医学研究、诊断试剂的开发和药物筛选等方面。
96孔板基因筛选方法

96孔板基因筛选方法96孔板基因筛选方法一:构建基因文库二:设计特异性引物三:96孔板基因筛选1. 实验前准备:高压灭菌(12连孔PCR小管--插在96孔黄色板上构成所需96孔板, 离心管250ul, 500ul, 1000ul各灭菌若干瓶, 枪头各种型号各灭菌若干盒. 固体.液体.上层培养基共1L, 液体培养基以25ml分装20瓶, 上层培养基按50ml加0.25g琼脂粉分装2瓶, 剩余作为固体培养基按100ml加1.5g琼脂粉分装. 配1M MgSO4. 20%麦芽糖各100ul 然后抽滤后用1.5ml 离心管分装. 无菌水(可用娃哈哈纯净水替代)用1.5ml 离心管分装后, 装瓶高压灭菌. 再灭一瓶无菌水.2. 培养XL-Blue E.coli, 大肠杆菌取一瓶灭菌过的液体培养基, 按25ml体系, 加入25ul tet抗生素(50mg/ml), 再加入250ul 1M MgSO4, 250ul 20%麦芽糖, 用接种针挑单克隆菌落于培养基中. 置于37℃摇床150转速过夜.3. 做第一批96孔板3.1 取1至多ul (确保含有该基因)所克隆基因的cDNA文库, 加上400ul 培养的XL-Blue大肠杆菌, 37℃静止30min3.2 将96孔底板放置于超净台上, 再将12连孔PCR小管, 插在该底板上做成所需的96孔板. 取一瓶灭菌的液体培养基(加入25ul tet抗生素, 250ul 1M MgSO4, 250ul 20%麦芽糖), 以150ul分装到96孔板每个孔中.3.3 30min后, 将第一步的混液按4ul 分装到96孔板每个孔中. 用保鲜膜包好96孔板,然后置于37℃摇床上200rpm培养8-10h.3.4 做PCR检测共3 次PCR3.4.1 每行做PCR找到含有该基因的具体某一行做8行PCR: 将每一行的12孔, 每孔各取5-10ul混匀. 这样做8行的各行的混液, 取这些混液1-2ul 做PCR. 电泳检测后发现哪孔有目标条带, 就表示相对应的那行有我们所要克隆的基因3.4.2 以上步找到的含有目标基因的行, 对该行12孔每孔做PCR 检测找到阳性单孔3.4.3 做浓度梯度PCR取10-20ul 阳性孔的菌液, 混匀后取1ul+ 99ul纯净水稀释100倍, 再取稀释100倍的混液10ul + 90ul 纯净水后就是稀释1000倍, 以此类推可以稀释10000倍.取稀释100倍的混液, 1ul, 3ul ,5ul PCR取稀释1000倍的混液, 1ul, 3ul ,5ul PCR取稀释10000倍的混液, 1ul, 3ul ,5ul PCR找到最低能够PCR检测出该基因的浓度4. 做第二批96孔板4.1 取1ul上步的最低浓度, 加上400ul 培养的XL-Blue大肠杆菌, 37℃静止30min4.2 将96孔底板放置于超净台上, 再将12连孔PCR小管, 插在该底板上做成所需的96孔板. 取一瓶灭菌的液体培养基(加入25ul tet抗生素, 250ul 1M MgSO4, 250ul 20%麦芽糖), 以150ul分装到96孔板每个孔中.4.3 30min后, 将第一步的混液按4ul 分装到96孔板每个孔中. 用保鲜膜包好96孔板,然后置于37℃摇床上200转培养8-10h.4.4 做PCR检测共3 次PCR4.4.1 每行PCR找到含有该基因的具体某一行做8行PCR: 将每一行的12孔,每孔各取5-10ul振荡混匀. 这样做8行的各行的混液, 取这些混液1-2ul 做PCR. 电泳检测后发现哪孔有目标条带, 就表示相对应的那行有我们所要克隆的基因4.4.2 以上步找到的含有目标基因的行, 对该行12孔每孔做PCR 检测找到阳性单孔4.4.3 做浓度梯度PCR取10-20ul 阳性孔的菌液, 混匀后取1ul+ 99ul纯净水稀释100倍, 再取稀释100倍的混液10ul + 90ul 纯净水后就是稀释1000倍, 以此类推可以稀释10000倍.取稀释100倍的混液, 1ul, 3ul ,5ul PCR取稀释1000倍的混液, 1ul, 3ul ,5ul PCR取稀释10000倍的混液, 1ul, 3ul ,5ul PCR找到最低能够PCR检测出该基因的浓度5. 重复做第二批96孔板的方法6. 取第5步找到的阳性孔, 进行铺板长出噬菌斑, 挑单个噬菌斑检测出阳性噬菌斑6.1 培养XL-Blue大肠杆菌过夜6.2 取10-20ul 阳性孔的菌液振荡混匀后, 先稀释100倍, 再稀释1000倍, 10000倍.6.3 在2灭菌过的1.5ml离心管中加入1ml 大肠杆菌, 6000rpm 离心1min. 去上清, 加入500ul 无菌水重悬, 再加入10ul 1M MgSO46.4 取1000倍和10000倍的稀释液各10ul 分别加入上步骤的离心管中, 轻微振荡混匀后, 于37℃培养箱中, 静置15min6.5 在这15min时间内准备LB 空白平板, 即将开始准备的固体培养基(按100ml加入1ml 1M MgSO4)融化后倒平板. 同时将开始准备的上层培养基(按50ml加入0.5ml 1M MgSO4)融化. 一个加入大半热水的三角瓶作温浴用.6.6 等15min结束后, 将离心管中的混液加入到10ml的离心管中(该10ml离心管放置于上步温浴三角瓶中), 并且加入3-4ml的上层培养基(温度大约50-60℃, 自己感觉上层培养基的瓶子握在手里较热, 但不至于很烫而拿不住) 很快混匀一下后, 倒入准备的平板上. 待很快凝固后, 用封口膜封好, 放置于37℃培养箱中培养过夜.6.7 挑单个噬菌板于100ul phage buffer 中, 充分振荡混匀后取1-2ul 进行PCR反应,剩余放4℃冰箱保存. 共挑10个或20个单噬菌斑.6.8 电泳后检测发现有目标条带的孔, 相对应的该噬菌斑便是我们所要找的含有目标基因的噬菌体.如未找到, 则再挑几个单噬菌斑, 如还没找到阳性克隆, 则重复第五步骤, 再次做96孔板. 然后找到含阳性克隆孔, 再进行铺板. 挑噬菌斑检测, 找到含有所克隆基因的噬菌体7. 将上步保存的含目标基因的噬菌体与phage buffer的混液, 转入BM大肠杆菌中使噬菌体脱臂成质粒7.1 接种: 取一瓶灭菌过的液体培养基, 按25ml体系, 加入25ul 氯霉素(50mg/ml), 25ul卡那霉素(50mg/ml). 用接种针挑单克隆BM25.8大肠杆菌于液体培养基中31℃过夜培养.7.2 取200ul培养的BM25.8大肠杆菌于1.5ml离心管中, 加入2ul MgCL2, 再加入第6步保存的含有目标基因的phage buffer混液, 31℃静置30min.7.3 在上步的离心管中加入300 灭菌过的LB液体培养基, 31℃摇床上200rpm 1h.7.4 其间准备平板, 将开始准备的固体培养基加热融解后, 待温度稍冷却但不凝固的时候, 按100ml体系, 加入100ul 氯霉素(50mg/ml), 100ul 卡那霉素(50mg/ml), 100ul carb 羧苄(50mg/ml). 混匀后倒平板.7.5 取7.3步的混液用涂玻棒涂板, 做浓度铺板, 取3 –5ul铺一板, 10–20ul铺一板(确保长出单菌落), 31℃培养箱培养过夜.7.6 在灭菌的1.5ml离心管中加入灭菌LB[按25ml体系, 加入25ul 氯霉素(50mg/ml),25ul 卡那霉素(50mg/ml)] 1 ml. 再挑上步培养出的单菌落于其中, 31℃摇床上200rpm 培养6-8 h .7.7 将培养的菌液提取质粒, 然后进行PCR鉴定, 以原先的特异引物和Triple引物分别为引物做PCR.7.8 以特异引物为引物的结果为设计的片段长度, 以Triple引物的结果为该基因的长度.。
2.DNA测序反应优化

Thank You !
29
7
大分子量测序 20 µL 96孔板
测序反应
DNA
1 µL
BigDye 引物 (3.2 pmol / µl) 去离子灭菌水
8 µL 1 µL 10 µL 总体积 20 µL
测序PCR循环条件
95 °C 5 min → (96 °C 30 sec → 50-55 °C 10 sec → 60 °C 4 min) x 50个循环 → 4 °C保温
9
测序产物纯化 10 µL 384孔板
酒精/EDTA/NaAc法
每管加入1 µL 125 mM EDTA,1 µL 3 M NaAc,到管底; 每管加入25 µL 100% 酒精,铝箔封严密,震荡混匀4次, 室温放置15 min; 2000-3000 x g离心30 min,立即倒置384孔板离心,至185 x g 停止,去除酒精; 每管加入35 µL 70% 酒精,1650 x g 4 °C离心15 min, 立即倒置384孔板离心,至185 x g停止,去除酒精; 重复70% 酒精洗涤1次; 让残余的酒精在室温挥发干,加入10 µL Hi-Di甲酰胺, 95 °C变性4 min,迅速冰冷4 min ,上样电泳。
150-300 ng
0.5-1 µg 2-3 µg
引物与模板的平衡最关键
质粒
200 ng : 3.2 pmol
PCR产物
☯ 100-200 bp ☯ 200-500 bp ☯ 500-1000 bp
1-3 ng : 3.2 pmol 3-10 ng : 3.2 pmol 5-20 ng : 3.2 pmol
分子越小,上样越快
盐离子、RNA、蛋白质等与 DNA片段竞争