腺病毒转染过程
腺病毒操作手册

Shanghai GeneChem Co. Ltd 重组腺病毒操作手册(Version 2.0)上海吉凯基因技术有限公司 二〇〇九年二月地址:上海张江高科技园区蔡伦路 720 弄 1 号 408 室 电话:86-21-51320189 网址: 邮编:201203 传真:86-21-51320179 Email:service@Page 1 of 4 Shanghai GeneChem Co. Ltd 重组腺病毒实验系统腺病毒是一种的线型双链 DNA 无包膜病毒, 它的 DNA 和核心蛋白形成的内核, 蛋白外壳 是直径约 80nm 的正二十面体,由 240 个六 面体和 12 个位于正二十面体顶部的五面体 构成。
Genechem 重组腺病毒系统是将携带外源基 因的腺病毒穿梭质粒与携带腺病毒基因组的 包装质粒共转染 HEK293 细胞, 通过 Cre/loxP 系统的作用实现重组,产生重组腺病毒。
重组腺病毒能够表达较大的外源基因片段, 感染分裂和非分裂细胞,具有广泛的宿主。
GFP-adenovirus 感染的代表细胞――腺病毒可以感染绝大多数细胞TCA8113SGC-7901NRKPC-3Huh-7GliaRabbit Mesenchymal Stem CellsHuman Mesenchymal Stem CellRat Cardiac Fibroblasts地址:上海张江高科技园区蔡伦路 720 弄 1 号 408 室 电话:86-21-51320189 网址: 邮编:201203 传真:86-21-51320179 Email:service@Page 2 of 4 Shanghai GeneChem Co. Ltd 重组腺病毒操作过程一, 注意事项:1. 操作病毒时请尽量使用生物安全柜。
腺病毒

• 腺病毒简介 • 人体腺病毒已知有52种,分别命名为adl~ad52,研究得最详细是 ad2。腺病毒基因组转录产生mRNA,已知的转录单位至少有5个:EⅠ区 位于病毒基因组左侧,可再分成EⅠA和EⅠB,与细 • • 腺病毒 • 胞转化有关;EⅡ区编码DNA结合蛋白,参与病毒的复制;EⅢ区编码出现 在宿主细胞表面的一种糖蛋白;EⅣ区位于ad2基因组右端,受EⅡ区编码 的DNA结合蛋白质调控;第5个转录单位在病毒感染中期合成ad2蛋白质Ⅳ。 • 腺病毒对啮齿类动物有致癌能力,或能转化体外培养的啮齿类动物细胞。 使细胞转化只需要腺病毒基因组的一部分,这些基因位于基因组的左端,约 占整个基因组的7%~10%。尽管腺病毒分布很广,但对人体不出现致癌性。 人体细胞是一类允许细胞(permissive cell),即这类细胞允许感染入侵的 病毒在细胞内复制增殖,最后细胞裂解死亡而释放出大量子代病毒。在体外 培养的多种人体肿瘤细胞中均未查出腺病毒颗粒,但在人的1号染色体上有 adl2的整合位点,这意味着人体细胞对于腺病毒也可能是非允许细胞,即 这类细胞在病毒感染后,病毒不能在细胞内复制增殖,但可整合在受感染细 胞的基因组内。这些细胞被病毒转化,表型发生改变,且可在体外无限期地 培养传代。
腺病毒
保定市第三中心医院 崔广奇
• 科技名词定义 • adenovirus • 一种具双链DNA的动物病毒。基因组大小 约为36 kb,常用于研究DNA复制、转录 和作为基因工程载体。 •
• 腺病毒 • 腺病毒(adenovirus)是一种没有包膜的直 径为70~90 nm的颗粒,由252个壳粒呈 廿面体排列构成。每个壳粒的直径为7~9 nm。衣壳里是线状双链DNA分子,约含 35 000 bp,两端各有长约100 bp的反 向重复序列。由于每条DNA链的5'端同相 对分子质量为55X103Da的蛋白质分子共 价结合,可以出现双链DNA的环状结构。
转录过程中慢病毒,腺病毒,逆转录病毒三种病毒表达系统的区别
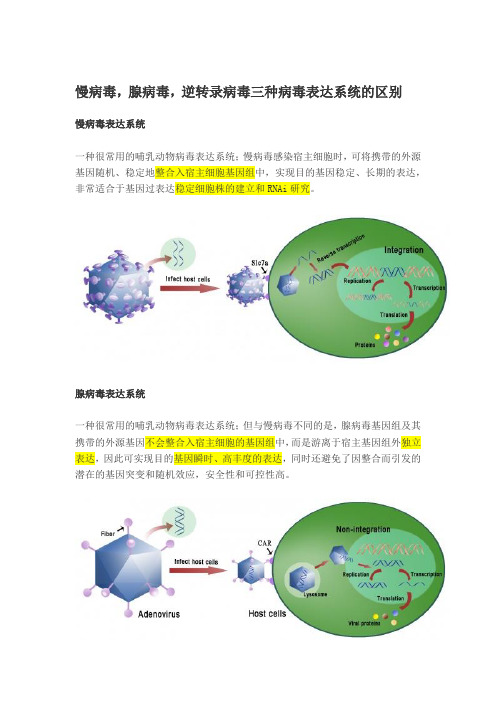
慢病毒,腺病毒,逆转录病毒三种病毒表达系统的区别
慢病毒表达系统
一种很常用的哺乳动物病毒表达系统;慢病毒感染宿主细胞时,可将携带的外源基因随机、稳定地整合入宿主细胞基因组中,实现目的基因稳定、长期的表达,非常适合于基因过表达稳定细胞株的建立和RNAi研究。
腺病毒表达系统
一种很常用的哺乳动物病毒表达系统;但与慢病毒不同的是,腺病毒基因组及其携带的外源基因不会整合入宿主细胞的基因组中,而是游离于宿主基因组外独立表达,因此可实现目的基因瞬时、高丰度的表达,同时还避免了因整合而引发的潜在的基因突变和随机效应,安全性和可控性高。
逆转录病毒表达系统
逆转录病毒将自身基因组及其携带的外源基因随机、稳定地整合入宿主细胞基因组中,可实现目的基因稳定、长期的表达逆转录病毒表达系统根据不同的包装细胞系可分为单嗜性、双嗜性和泛嗜性,不同包装细胞产生的逆转录病毒能够特异性地感染某一个或某一类宿主细胞。
辉骏生物采用的病毒包装细胞系为plat E 细胞系,该细胞系包含gag、pol和env基因,因此转染单一表达质粒就可以包装出逆转录病毒,但是该病毒具有很强的针对性,只适用于大鼠、小鼠细胞稳定株的建立。
三种病毒表达系统的特点。
腺病毒中文操作手册

腺病毒载体操作手册中文版腺病毒重组系统AdEasyTM操作手册目录第一章简介 1第二章应用重组腺病毒的优点 2第三章AdEasyTM 技术 33.1 技术概况33。
2 AdEasyTM系统中产生重组腺病毒的时程3 第四章主要流程 44。
1 将基因克隆入AdEasyTM转移载体44.1。
1 克隆的一般原则44。
1.2 构建重组AdEasyTM转移载体54.2 细菌内AdEasyTM重组子的产生54。
2.1 共转化的一般原则54。
2。
2 共转化方法54.2.3 预期结果54.3 AdEasyTM重组质粒的筛选和扩增64。
4 AdEasyTM重组子转染QBI—293A细胞6 4.4。
1 细胞铺板64。
4。
2 磷酸钙转化技术7第五章常用技术85。
1 QBI-293A细胞培养85.1。
1 QBI—293A细胞的初始培养85。
1.2 QBI—293A细胞的维持培养和增殖8 5。
1.3 QBI-293A细胞的冻存85.2 QBI-293A细胞的转染和病毒空斑的产生9 5.2。
1 感染QBI—293A细胞95。
2.2 病毒空斑形成95.2。
3 琼脂糖覆盖被感染细胞95.3 MOI测定105.4 腺病毒感染力测定105。
4.1 X-Gal染色11 5。
5 重组腺病毒的筛选和纯化115。
5.1 挑选最佳重组腺病毒:表达和基因输送11 5.5。
2 病毒空斑挑选和小量扩增125.5。
3 Western杂交135.5.4 Southern杂交和点杂交135.5.5 病毒裂解产物PCR 145。
5。
6 免疫测定145。
5。
7 功能测定145.6 病毒颗粒在QBI—293A细胞中的大量扩增15 5.7 两次氯化铯密度梯度离心纯化重组腺病毒16 5。
7.1 不连续密度梯度离心175.7。
2 连续密度梯度离心175.7。
3 病毒溶液去盐和浓集175。
8 病毒滴度测定185。
8。
1 O。
D。
260 nm (VP/ml) 195。
8。
腺病毒

生活周期
腺病毒的生活周期可以分为两个截然不同却又不能割裂开来的两个阶段。第一阶段包括腺病毒颗粒粘附和进 入宿主细胞,将基因组释放到宿主细胞核中,以及有选择性地转录和翻译早期基因。在这个阶段,细胞为病毒基 因组复制和腺病毒晚期基因表达并最终释放成熟的感染颗粒,即第二阶段,作好了准备。第一阶段将在6~8个小 时内完成,第二阶段则更快,只需4~6个小时。
分类
分类及自上个世纪50年代发现并成功分离腺病毒以来,已陆续发现了100余个血清型,其中人腺病毒有52种, 分为A、B、C、D、E和F六个亚群(subgroup)。基因治疗常用的人的2型及5型腺病毒在血清学分类上均属C亚群, 在DNA序列上有95%的同源性。二者的增殖能力非常强,滴度通常可以达到109pfu (plaque forming unit)/ml, 其在单个细胞中的基因组拷贝数可达104(约占细胞总DNA的10%)。病毒颗粒比较稳定,通过CsCl梯度离心可以 达到1010~1011pfu/ml,满足动物实验的要求。
防治原则
腺病毒的甲醛灭活疫苗已被用于某些人群的预防,而且将来有被用人二倍体细胞培养的减毒活疫苗所替代的 可能。但因腺病毒对动物具有致癌作用,人们对全病毒疫苗的作用与安全性存有疑虑。此外加强游泳池和浴池水 的消毒,可使水传播性结膜炎爆发的危险性降至最小,在作眼的检查时应严格无菌操作,对所用设备充分灭菌, 也可控制流行性结膜炎的发生。对腺病毒感染的治疗仍无有效药物。
许多腺病毒在肠道细胞中复制,随粪便排出,但大多血清型与胃肠道疾病无关。而40型和41型腺病毒可引起 婴幼儿与年少(4岁以下)儿童的胃肠炎,致腹痛、腹泻。C组腺病毒能引起某些婴幼儿肠套叠。
腺病毒11、12型能引起儿童急性出血性膀胱炎,尿中出现病毒。37型可引起女性宫颈炎和男性尿道炎,常由 性传播感染。在免疫功能低下者可引起偶发或严重的病毒感染,尤其在器官移植病人中发生严重呼吸道感染和病 毒性肝炎,多由1、5和7型腺病毒引起。艾滋病患者可感染多种血清型腺病毒,并能出现抗原性介于中间的杂合 型毒株,而且常为致死性腺病毒感染。主要原因是腺病毒的E1A蛋白可反式激活HIV的转录,加速HIV的复制。临 床发现37%的艾滋病患者病毒性腹泻是由腺病毒所致。
腺病毒的包装与制备

腺病毒的包装与制备1 转染AD293第一轮:1. pAd质粒转染AD293,可使用六孔板或60mm皿。
当细胞出现明显细胞病变效应(CPE)时回收细胞,(吸去培养基用PBS洗一遍,若细胞已经悬浮则取所有培养基低速离心,细胞用PBS洗一遍),并用0.5ml PBS重悬。
置于1.5ml microcentrifuge tube中2. 裂解细胞:进行四轮-80?/37?3. 12000g 10min 室温离心,将上清移入新的离心管中。
-80?保存。
第二轮:1. 取第一轮的病毒液50μl(或设置梯度)感染100mm皿。
(如细胞第一天就出现病变则用量太高,一般应在第一天少量病变,第二三天全部病变)。
重复第一轮方法裂解细胞并收集腺病毒,终体积1ml第三轮取第二轮的病毒感染4个150mm皿(100μl/皿或设置梯度),并用CsCl密度梯度法纯化腺病毒。
2 透析Transfecting AD-293 Cells以Lipofectamine? 2000说明书为本,更改了细胞密度和DNA量51. One day before transfection, plate 4x 10 cells per 6-wellofgrowth medium without antibiotics so that cells will be 50-70% confluent at the time of transfection. 2. For each transfection sample, prepare complexes as follows:a. Dilute 4μg linearized DNA in 250 μl of Opti-MEM I Reduced Serum Medium without serum (or other medium without serum). Mix gently.b. Mix Lipofectamine? 2000 gently before use, then dilute 10μl lipofectaminein250 μl of Opti-MEM I Medium. Incubate for 5 minutes at room temperature. Note:Proceed to Step c within 25 minutes.c. After the 5 minute incubation, combine the diluted DNA withdilutedLipofectamine? 2000 (total volume = 100 μl). Mix gently andincubate for 20 minutes at room temperature (solution may appear cloudy). Note:Complexes are stable for 6 hours at room temperature. 3. Add the 500 μl of complexes to each well containing cells and medium. Mix gently by rocking the plate back and forth.4. Incubate cells at 37?C in a CO2 incubator for 17-10days prior to testing for GFP. Medium may be changed after 4-6 hours.Preparing the Primary Viral Stocks1. Prepare a small dry ice-methanol bath and a small 37?C water bath and placethem in the laminar flow hood.2. Carefully remove growth medium from adenovirus-producing AD-293 plates andwash the cells once with PBS. Take care not to lose any clusters of floating andpartially attached cells during this process.Note :If the cells are already mostly detached, pipet up and down gently in the growth medium until cells become completely resuspended. Transfer cell suspension to a screw cap centrifuge tube and pellet the cells by low speed centrifugation. Aspirate medium, and wash the cells once with 0.5 ml of sterile PBS. Resuspend the cell pellet in a fresh 0.5 ml sterile PBS (per 60-mM dish) and proceed to Step 5. 3. Add 0.5 ml of PBS to each plate of cells to be harvested. Collect the cells by holding the plate at an angle and scraping the cells into the pool of PBS with a cell lifter.4. Transfer the cell suspension to a 1.7-ml screw-capmicrocentrifuge tube. If duplicate DNA samples were transfected, the cells from duplicate samples may be combined in the microcentrifuge tube at this stage.5. Subject the cell suspension to four rounds of freeze/thaw by alternating the tubes between the dry ice-methanol bath and the 37?C water bath,vortexing briefly after each thaw.Note :Each freeze and each thaw will require approximately 5 minutes’ incubationtime.6. Collect cellular debris by microcentrifugation at 12,000 × g for 10 minutes at room temperature.7. Transfer the supernatant (primary virus stock) to a fresh screw-cap microcentrifuge tube. Viral stocks can be stored for more than one year at –80?C.腺病毒扩增1. Plate 5 X 106 QBI-293A cells in a 100 mm culture dish in 10 mL DMEM 5%.2. Remove 50μl(100μl) of the viral stock of the first amplification, complete to 1 mL with DMEM 5% and mix. This dilution will give a MOI of about 5.3. Remove the medium from the 100 mm dish, delicately add the viral particle mix onto the cells taking care not to disturb the monolayer and spread by slowly rocking the dish 3 times in a cross shape. Incubate for 90 minutes at 37?C in 5% CO2.4. Add 9 mL of DMEM 5%.5. Incubate at 37?C in a CO2 incubator for 72 hours. At this point you should have between 5 x91010 and 5 x 10 VP in 10 mL of DMEM 5%. Perform a MOI test (section 5.3) to estimate the titer if desired.If this quantity of virus is sufficient, you can immediately proceed to the titration step (section 5.8). In this case, collect the cells in a centrifuge tube, spin at 600 x g for 5 min, discard supernatant and resuspend the cell pellet in a minimal volume, typically 1:10 of the original volume or about 1mL. Then proceed with 3 freeze/thaw cycles (-20?C / 37?C), spin at maximum speed on a tabletop centrifuge to remove cellular debris, and collect the supernatant. Titer as described in section 5.8. 6. Perform three freeze/thaw cycles at -20?C until completely frozen/37?C until fully thawed. 7. Pellet cell debris in a sterile 15 mL conical tube. Centrifuge at maximum speed in a tabletop centrifuge for 10 minutes, remove and store the supernatant in another sterile 15 mL conical tube at -20?C or -80?C.8. Plate 3 x 107 QBI-293A cells in 3 x 175 cm2(150mm dish) culture flasks (1 X 107cells/flask).9. Mix 3 mL of cell lysate supernatant from step 7 with 12 mL DMEM 5%. Remove the mediumfrom the flask, then delicately add 5 mL of supernatant mix perflask on the cells, taking care notto disturb the monolayer, and spread by slowly rocking the dish 3 times in a cross shape. Incubate for 90 minutes at 37?C in 5% CO2. This should give a MOI of 25.10. Complete to 30 mL/flask with DMEM 5%.11. Incubate at 37?C in a CO2 incubator for 48 to 72 hours. At this point you should have between 3 x 1010 and 3 x 1011 VP in 90mL of DMEM 5%. Perform a MOI test (section 5.3) to estimate the titer if desired. Again, if this quantity of virus is sufficient, you can immediately proceed to the titration step (section 5.8). In this case, collect the cells in a centrifuge tube, spin at 600 x g for 5 min, discard supernatant and resuspend the cell pellet in a minimal volume, typically 1:10 of the original volume. Then proceed with 3 freeze/thaw cycles (-20?C / 37?C), spin at maximum speed on a tabletop centrifuge to remove cellular debris, and collect the supernatant. Titer as described in section 5.8.12. Perform three freeze/thaw cycles at -20?C/37?C.13. Pellet cell debris in a sterile 50 mL conical tube. Centrifugeat maximum speed in a tabletop centrifuge for 10 minutes. Remove and store the supernatant.14. Plate 3 x 108 QBI-293A cells in 30 x 175 cm2 culture flasks (1 X 107 cells/flask). 15. Mix 45 mL of cell lysate supernatant from step 13 with 105 mL DMEM 5%. Remove the medium from the 175 cm2 flasks, pour 5 mL of the cell lysate mix per flask on the cells, taking care not to disturb the cell monolayer, and spread by slowly rocking the flask 3 times in a cross shape. Incubate for 90 minutes at 37?C in 5% CO2. This should give a MOI of 25.16. Complete to 30 mL/flask with DMEM 5%.17. Put the cells back at 37?C in a CO2 incubator for 2-3 days. You should now have between 3 x 111210 to 3 x 10 VP. Perform a MOI test (section 5.3) to estimate the titer if desired. If you want to produce viral particles, first collect and pellet the infected cells, then resuspend in 5 mL DMEM 5%. Extract viral particles by performing three freeze/thaw cycles at -20?C/37?C; pellet cell debris by centrifugation. At this point you should have between 3 x 1011 to 3 x 1012 recombinant virus particles in 5 mL DMEM 5%, for a final VP/mL of 6 X 1010 to 6 x 1011. Your viral particle pre-stock will now be ready to be titrated directly, asdescribed in section 5.8, or purified, using standard cesiumchloride gradients (section 5.7). Further amplifying your recombinant virus on 109 cells will result in the production of a viral stock, while amplification on 1 x 1010 cells is technically a viral production. You should never use the virus from a production stock to infect additional cells because you will at the same time significantly increase the amount of RCA generated. Instead, use virus from an earlieramplification steps such as the stock, pre-stock or pre-amplification in order to produce more virus particles. You can eventually go back to the purified eluted plaque in order to minimize RCAproduction as much as possible. If a purified eluted plaque is no longer available, you will have to perform a plaque assay of your recombinant virus and start the amplification step again from a purified eluted plaque, after analysis of your clone. If more material than produced in the first 4 passages(see Table 5) is needed, remember to always return to the previous passage as your source for virus. For example, use virus from passage 2 in order to generate passage 3. Note that once you have depleted all of your viruses from passage 2, you must use virus from the first amplification in order to create a new passage 2. Proceeding in this fashion will keep the level of RCA as low as possible. If you want to overexpress a protein, pellet the cells and extract the protein according to its characteristics. Typically, about 1-5 mg of a well-expressed protein can beretrieved from the pellet. It is recommended to first determine the best time pi to extract the protein on a smallscale, then to proceed with large-scale protein production.CsCl密度梯度法纯化腺病毒1 Add either purified Ad or unpurified Ad (medium) to monolayer culture cells (50-100pfu/cell)2 If using one T150 flask total 10-12 ml medium is used to cover the cells and allow cells to culture for one day3 Cells should look swollen and part of the cells may be floating. Another 10ml of complete medium is added into cells and allow another day culture (36-40hrs infection period )4 All of the cells should be floating. Collect all the cells and resuspend them in 0.5-1ml of complete medium . Also save the culture medium and store at -70?.5 Freeze in methanol/dry ice bath and thaw at 37?. Repeat for 3-4times6 Spin at high speed for 5min.7 Make 40% and 15% CsCl in TBS, PH8.1(50ml each and keep at 4?)8 Make CsCl gradient solution(5ml of 15% and 4.5ml of 40%) in Beckman centrifuge tube (14*89mm, which frist sw41 rotor)9 Load the supernatant from step 6 on the top of gradient10 Centrifuge at 4? 30000rpm for 16hr11 Two bands can be seen: the faint top band (mainly defective Ad) and the lower Ad band. Only the lower band is collected.12 the Ad is dialyzed in TBS PH8.1 for 1hr and then in TBS containing 10% glycol twice (1hr each time)13 Determine the Ad concentration, aliquot into microfuge tube(50μl each )and store at -70?TBS: tris 10mM, NaCl 0.9%, PH8.115% CsCl(50ml): 9.085g CsCl+47.69ml TBS40% CsCl(50ml): 28.45g CsCl+42.7ml TBSDialyze:1 透析袋: MW 8000-144002 透析缓冲液: TBS PH8.1 灭菌甘油步骤:1冲洗透析袋,如果是蛋白则需NaHCO-NaEDTA煮沸处理后用蒸馏水洗。
(整理)腺病毒载体操作手册1407-R2

腺病毒载体操作手册一、实验流程制备腺病毒穿梭质粒,分别高纯度无内毒素抽提腺病毒穿梭质粒和骨架质粒,共转染293A细胞,转染后6h更换为完全培养基,培养十几天,在中间四五天左右更换一次新鲜培养基,然后收集细胞和1ml培液置于15ml离心管后,液氮/37度冻融三次(冻-融要彻底),2000rpm离心5分钟,取上清即为病毒液初代原液。
连续三代反复扩增收集病毒后,行病毒的大量扩增,然后通过CsCl密度梯度离心-透析联用法纯化病毒。
二、实验材料(一)腺病毒载体、包装细胞和菌株该病毒包装系统为两质粒系统,组成为穿梭质粒(包括pHBAd-CMV-IRES-GFP,pHBAd-CMV-IRES-RFP,pHBAd-U6-GFP, pHBAd-U6-RFP)和骨架质粒pBHGlox(delta)E1,3Cre。
其中穿梭质粒pHBAd-CMV-IRES-GFP和pHBAd-U6-GFP能表达绿色荧光蛋白(GFP)。
pHBAd-CMV-IRES-RFP和pHBAd-U6-RFP能表达红色荧光蛋白(RFP)。
1、载体信息1) 腺病毒克隆载体图谱如下:各载体用途如下表:2)骨架质粒信息如下:2、细胞株293A,腺病毒的包装细胞,为贴壁依赖型成上皮样细胞,经培养生长增殖形成单层细胞,生长培养基为DMEM(含10% FBS)。
3、菌株大肠杆菌菌株DH5α。
用于扩增腺病毒载体和腺病毒骨架载体质粒。
三、包装细胞293A细胞的培养(一)293A细胞的冻存293A细胞来源于一个用作空斑测定的亚克隆,具有易使用和易转染的特性。
该细胞株对于高细胞密度很敏感,当细胞超过70%汇合时,一些细胞可能会丢失它们的表型。
若细胞密度持续在70%以下,QBI-293A细胞则能连续培养3~4个月维持原有细胞特性。
若以购买得到的293A作为第一代,则30代内能得到最佳结果。
随着传代的次数增加,293A细胞会出现生长状态下降、突变等。
为了防止此类现象的出现,我们需要在开始就对细胞进行大量冻存,以保证实验的稳定性和持续性。
腺病毒转染细胞步骤及方法

腺病毒转染细胞步骤及方法腺病毒(Adenovirus,ADV)是一种线性双链DNA 病毒。
腺病毒不仅可以作为哺乳动物基因转移载体而得到广泛应用,亦可被用来在人体细胞中过量表达蛋白(Massie et al., 1998a,b)。
迄今为止已有大量关于腺病毒及其应用的综述表明:重组腺病毒在科学研究和治疗应用领域都具有很大的潜力,同时作为基因转移工具相对于其他病毒载体有多方面的优势。
运用了去除5' ITR、包装信号和E1 序列的全新的腺病毒骨架基因,无需进行细菌体内的同源重组步骤,无需使用多蚀斑分离方法分离目的重组腺病毒的步骤,因此较其他系统生长更快,并且大大降低了重组形成野生型可复制腺病毒的危险;无E1a,不产生复制型腺病毒,因此更为安全。
二、腺病毒载体优势1)可感染的细胞种类多,宿主范围广,几乎可以感染所有类型的细胞;2)感染效率高,重组腺病毒被广泛应用于使用脂质体转染效率较低细胞的基因转导和蛋白表达;3)包装外源基因的片段大,高达8kb;4)腺病毒载体构建及包装容易操作,易于制备出滴度高的大量病毒;5)当腺病毒感染细胞后,病毒基因组存在于细胞染色体外,不整合到细胞染色体中,避免了因整合引起的基因突变及激活致癌基因,生物安全性高。
三、流程描述制备腺病毒颗粒的穿梭质粒及其骨架质粒,上述质粒载体分别进行高纯度无内毒素抽提,用转染试剂RNAi-Mate 进行共转染293A 细胞,转3染6小时后更换为完全培养基,培养7-15 天,每3天左右补充一次新鲜培养基,然后收集细胞和上清液置于离心管中,冻融三次,4000 rpm 离心10 分钟,取上清即为病毒液初代原液。
连续三代反复扩增收集病毒后,腺病毒的大量扩增,而后对其纯化和浓缩后得到高滴度的腺病毒浓缩液,在293A 细胞中测定并标定病毒滴度。
在一定滴度范围内的腺病毒颗粒可以满足大部分体内体外实验需求。
四、具体步骤细胞培养活细胞计数用无血清培养基把细胞悬液稀释到200~2000 个/ml (一般稀释倍数为100 倍),在0.1 ml 的细胞悬液中加入0.1 ml 的0.4%的台盼蓝溶液。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
腺病毒转染过程
腺病毒感染细胞的过程是从腺病毒纤毛的头节区粘附到细胞表面的特异性受体开始的。
因为人腺病毒主要与柯萨奇B病毒共用一种受体,因此这种受体被称为柯萨奇/腺病毒受体即CAR(coxsackie/adenovirus receptor)。
接下来病毒纤毛基底部五邻体表面的三肽RGD与细胞表面的αvβ3和αvβ5整合素结合,通过内吞作用将腺病毒内化到细胞中并进入溶酶体。
在溶酶体的酸性环境下,腺病毒衣壳的构象将发生变化,被从溶酶体中释放出来,躲过溶媒体的消化作用。
最后,腺病毒颗粒转位到细胞核,通过核孔将病毒DNA释放到细胞核内。
相对于脂质体转染,腺病毒基因组进入细胞核是一个非常高效的过程,一般可以达到40%,前者虽然进入胞质的效率与后者相当,而DNA进入细胞核的效率却只有前者的1/1000。
一旦病毒基因组进入细胞核,就将进行一系列的复杂而有序的逐级放大的剪切和转录过程。
一般的,以病毒DNA开始复制为分界线,按转录时间的先后,将腺病毒基因大致区分为早期(E1~4)和晚期转录单位(L1~5)。
各种腺病毒基因又可以进一步地分为更小的转录单位,如E1区可以进一步分为E1A和E1B,每个转录单位都至少有一个独特的启动子。
腺病毒基因组进入细胞核后,细胞转录因子首先与E1A区上游的增强子结合,表达E1A蛋白,该蛋白的作用是调节细胞代谢,使病毒DNA更易于在细胞中复制。
E1A蛋白还可以激活其他早期基因(E1B、E2A、E2B、E3和E4)的启动子,其中E2B驱动另外三个与病毒复制有关的早期基因转录单位末端蛋白前体(pTP, precursor terminal protein)、单链DNA结合蛋白(ssDBP, single-stranded DNA binding proteins)以及DNA聚合酶(DNA pol, DNA polymerase)的表达,这三个基因的表达产物紧密地结合成一个复合物,与至少三种细胞蛋白相互作用,启动病毒基因组的复制。
一般而言,DNA的复制是由RNA启动的,而在腺病毒却是所谓的蛋白启动(protein-priming)。
如前所述,腺病毒双链DNA的每条单链的5′端有pTP蛋白结合,pTP通过其Ser-OH与DNA 5′端的dCMP 5′磷酸之间形成磷酸二脂键。
腺病毒的DNA复制首先是以5′端结合有pTP的dCMP作为引物,以3′端的末端反向重复序列(ITR)为模板,进行链置换(strand displacement)合成,置换出的单链分子可以自我退火环化,形成锅柄样环形分子,然后这种环形分子再以相同的机制合成出子代双链DNA分子。
病毒基因组复制通常在感染后数小时开始,同时早期基因的转录和翻译被关闭,晚期基因开始表达。
大部分的晚期基因的转录是以一个共同的主要晚期启动子(MLP, Major Late Promoter)调控的。
实际上,MLP的活性与病毒基因组复制密切相关,有研究表明一旦腺病毒基因组开始复制MLP的活性将明显增强,对此我们将另外行文详述。
晚期基因主要编码腺病毒的结构蛋白。
病毒结构蛋白在细胞核内聚集形成病毒衣壳,病毒的基因组被包装进去,形成有感染能力的病毒颗粒,并最终裂解宿主细胞被释放出去,完成腺病毒的生活周期。
腺病毒有明显的种属特异性,人的野生型5型腺病毒(wtAd5)感染其他的非人类细胞(如鼠类细胞)后可以表达早期基因,基因组也可有一定程度的复制并能够形成一些不成熟的病毒颗粒,却不能形成成熟的病毒颗粒,也不能二次感染其他细胞。