反相高效液相色谱法测定环境有害物质酚酞含量的研究2012
通则0512高效液相色谱法

高效液相色谱法:系采用高压输液泵将规定的流动相泵入装有填充剂的色谱柱,对供试品进行分离测定的色谱方法。
注入的供试品,由流动相带入色谱柱内,各组分在柱内被分离,并进入检测器检测,由积分仪或数据处理系统记录和处理色谱信号。
1.对仪器的一般要求和色谱条件高效液相色谱仪由高压输液泵、进样器、色谱柱、检测器、积分仪或数据处理系统组成。
色谱柱内径一般为3.9~4.6mm,填充剂粒径为3~10μm。
超高液相色谱仪:是适应小粒径(约2μm)填充剂的耐超高压、小进样量、低死体积、高灵敏度检测的高效液相色谱仪。
(1)色谱柱反相色谱柱:以键和非极性基团的载体为填充剂填充而成的色谱柱。
常见的载体有硅胶、聚合物复合硅胶和聚合物等;常用的填充剂优十八烷基硅烷键合硅胶、辛基硅烷键合硅胶和苯基键合硅胶等。
正相色谱柱:用硅胶填充剂,或键合极性基团的硅胶填充而成的色谱柱。
常见的填充剂有硅胶、氨基键合硅胶和氰基键合硅胶等。
氨基键合硅胶和氰基键合硅胶也可用作反向色谱。
离子交换色谱柱:用离子交换填充剂填充而成的色谱柱。
有阳离子交换色谱柱和阴离子交换色谱柱。
手性分离色谱柱:用手性填充剂填充而成的色谱柱。
色谱柱的内径和长度,填充剂的形状、粒径与粒径分布、孔径、表面积、键合基团的表面覆盖度、载体表面基团残留量,填充的致密与均匀程度等均影响色谱柱的性能,应根据被分离物质的性质来选择合适的色谱柱。
温度会影响分离效果,品种正文中未指明色谱柱温度时系指室温,应注意室温变化的影响。
为改善分离效果可适当提高色谱柱的温度,但一般不宜超过60℃。
残余硅羟基未封闭的硅胶色谱柱,流动相的pH值一般应在2~8之间。
残余硅羟基已封闭的硅胶、聚合物复合硅胶或聚合物色谱柱可耐受更广泛pH值的流动相,适合于pH值小于2或大于8的流动相。
(2)检测器最常用的检测器为紫外-可见分光检测器,包括二极管阵列检测器,其他常见的检测器有荧光检测器、蒸发光散射检测器、示差折光检测器、电化学检测器和质谱检测器等。
反相高效液相色谱法测定环境有害物质—酚酞含量的研究

1 实验 部分
1 1 仪 器 、 剂 与标准 品 . 试 美 国安捷 伦公 司 A in 20If i P C, get 6 nit H L 配 l 1 ny D D光 电二 极管 阵列 检测 器 。 A
K 50 D Q 2 0 B型超声 波仪 ( 山市 超 声 仪 器 有 限 昆
添加 了 1 %磷 酸 ( O ) 冲盐 , 的添 加 了 0 5 H P 缓 有 . %
公 司制造 ,0 HZ 功率 :o w) 4K , 2o 。
甲醇( P C色谱纯)德国默克公司 M r 制造。 HL , ek c
酚 酞标 准 品 , 国 D . he s r r 司制 造 , 德 r E rnt f 公 oe 纯
方 法开 发 的 中间步骤 可 以多设 几步 流 动相 组分
依次改变的细化条件来 实现逐步优化 , 现在 的方 向 是 : 次调 高 甲醇组 分 , 依 比较得 出最佳 色谱 峰形 。甲 醇 : = 0 :0 , 出的色 谱 图如 图 5 水 9 % 1% 得 :
图 3 文 献 中 酚酞 的标 准 紫 外 吸 收 光 谱 图
关键词 : 反相高效液相色谱法 ; 等度洗脱 ; 酚酞 ; 超声萃取
中图 分 类 号 : 6 7 7 0 5 . 文献标识码 : A
0 引 言
酚酞 被 欧盟化 学 品管理 局 ( C A) 为 高度 关 EH 列 注物 质 ( V S HC) 一 , 往 欧盟 的化 学 品 或 物 品 亡 之 销 F L 的 SH V C含 量 超 过 0 1 的 , 会 被 通 报 或 召 回。 .% 将
一
3 一
4% , 0 流动 相流 速 :mL m n 紫外 检 测 波长 A未知 , 1 / i,
反相高效液相色谱法检测丁苯酞原料药中的杂质
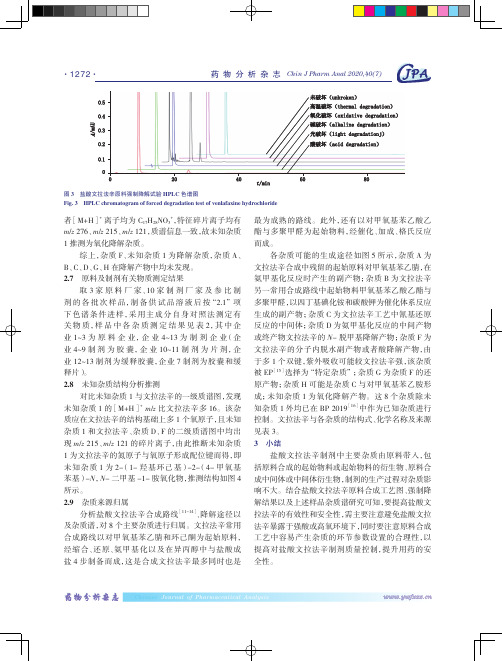
离子均为C17H28NO3+,特征碎片离子均有215、m/z 121,质谱信息一致,故未知杂质推测为氧化降解杂质。
杂质F、未知杂质1为降解杂质,杂质A、在降解产物中均未发现。
原料及制剂有关物质测定结果原料厂家、10家制剂厂家及参比制样品,制备供试品溶液后按“2.1”项下色谱条件进样,采用主成分自身对照法测定有品中各杂质测定结果见表2,其中企料企业,企业4~13为制剂企业(企为胶囊,企业10~11制剂为片剂,企制剂为缓释胶囊,企业7制剂为胶囊和缓未知杂质结构分析推测对比未知杂质1与文拉法辛的一级质谱图,发现的[M+H]+m/z比文拉法辛多16。
该杂质应在文拉法辛的结构基础上多1个氧原子,且未知和文拉法辛、杂质D、F的二级质谱图中均出/z 121的碎片离子,由此推断未知杂质为文拉法辛的氮原子与氧原子形成配位键而得,即为2-(1-羟基环己基)-2-(4-甲氧基二甲基-1-胺氧化物,推测结构如图4杂质来源归属分析盐酸文拉法辛合成路线[11-14]、降解途径以8个主要杂质进行归属。
文拉法辛常用最为成熟的路线。
此外,还有以对甲氧基苯乙酸乙酯与多聚甲醛为起始物料,经催化、加成、格氏反应而成。
各杂质可能的生成途径如图5所示,杂质文拉法辛合成中残留的起始原料对甲氧基苯乙腈,氨甲基化反应时产生的副产物;杂质B为文拉法辛另一常用合成路线中起始物料甲氧基苯乙酸乙酯与多聚甲醛,以四丁基碘化铵和碳酸钾为催化体系反应生成的副产物;杂质C为文拉法辛工艺中氰基还原反应的中间体;杂质D为氨甲基化反应的中间产物或终产物文拉法辛的N-脱甲基降解产物;杂质文拉法辛的分子内脱水副产物或者酸降解产物,于多1个双键,紫外吸收可能较文拉法辛强,被EP[15]选择为“特定杂质”;杂质G为杂质原产物;杂质H可能是杂质C与对甲氧基苯乙胺形成;未知杂质1为氧化降解产物。
这8个杂质除未知杂质1外均已在BP 2019[16]中作为已知杂质进行控制。
文拉法辛与各杂质的结构式、化学名称及来源见表3。
反相高效液相色谱法测定可乐、茶叶中的咖啡因

反相高效液相色谱法测定可乐、茶叶中的咖啡因
帅琴;龚宇;杨薇;王丽
【期刊名称】《分析试验室》
【年(卷),期】2002(21)1
【摘要】研究了反相高效液相色谱法 (RP HPLC)直接测定茶叶水提取物、可乐饮料中的咖啡因的新方法 ,采用PE C1 8柱 ,以CH3OH H2 O HAc( 30 :69 92 :0 0 8)溶液作流动相 ,紫外检测波长为 2 60nm。
可在 8min内将咖啡因与基体分离 ,咖啡因含量与峰面积在1 0 μg/mL~1 0 0 μg/mL范围呈线性关系 ,回归方程为 :Y
=335 67c( μg/mL) - 1 2 642 1 ,r=0 .9997。
该法可用于可乐饮料。
【总页数】3页(P68-70)
【关键词】咖啡因;HPLC;茶叶;可乐;反相高效液相色谱;质量监控;饮料;分析
【作者】帅琴;龚宇;杨薇;王丽
【作者单位】中国地质大学材料科学与化学工程学院
【正文语种】中文
【中图分类】TS275;TS207.3
【相关文献】
1.反相高效液相色谱法测定茶叶和速溶咖啡中咖啡因的含量 [J], 胡媛;周艳明;牛森;刘笑
2.反相高效液相色谱法测定茶叶和速溶咖啡中咖啡因的含量 [J], 胡媛;周艳明;牛森;刘笑
3.反相高效液相色谱法测定茶叶中的咖啡因 [J], 陈中道;朱军;王琳;孙建生
4.反相高效液相色谱法测定茶叶和速溶咖啡中咖啡因的含量 [J], 胡媛;周艳明;刘笑
5.反相高效液相色谱法测定可乐中咖啡因的含量 [J], 武开业
因版权原因,仅展示原文概要,查看原文内容请购买。
反相高效液相色谱法测定饮用水中的酚类化合物

反相高效液相色谱法测定饮用水中的酚类化合物
王碧云
【期刊名称】《福建分析测试》
【年(卷),期】2006(015)003
【摘要】介绍了反相高效液相色谱法测定饮用水中的酚类物质.该法采用的固相萃取预处理法操作简便,节省溶剂,且稳定性好,回收率高.实验以ODS柱为分离柱,采用梯度洗脱,可编程紫外检测器进行样品检测.方法准确,重现性好,杂质干扰少,检出限低.
【总页数】3页(P20-22)
【作者】王碧云
【作者单位】国家城市供水水质监测网海口监测站,海南,海口,570203
【正文语种】中文
【中图分类】O657.72
【相关文献】
1.固相萃取-反相高效液相色谱法测定水中五种酚类化合物 [J], 梁景华;霍柱堂
2.固相萃取-高效液相色谱法测定饮用水中酚类化合物 [J], 张丽;李楠;万延延
3.饮用水加氯消毒前后水中酚类化合物的形态改变及石英砂滤层对酚类化合物吸附作物的探讨 [J], 陈明
4.枪头式羧基化多壁碳纳米管固相萃取/高效液相色谱测定饮用水中双酚类化合物[J], 朱培杰;涂雪元;周家欢;陈奕全;刘艳清;汪洪武
5.应用连续流动分析仪一次在线蒸馏同时测定生活饮用水中的挥发酚类化合物和氰化物 [J], 殷爱琴;夏玉庆;肖丽;冯忠莲
因版权原因,仅展示原文概要,查看原文内容请购买。
RP—HPLC法测定酚酞片中酚酞的含量

RP—HPLC法测定酚酞片中酚酞的含量目的建立RP-HPLC法测定酚酞片的含量。
方法采用Agilent C18色谱柱(5 μm,4.6 mm×150 mm),柱温为25℃,以甲醇-0.05 mol/L NH4H2PO4(用磷酸调pH至3.31)(70:30)为流动相,流速为1 mL/min,检测波长为275 nm。
结果酚酞在(19.968~139.776)μg/mL的范围内线性关系良好(r = 0.9999,n = 5),测得平均回收率为100.87%,RSD = 0.88% (n = 9)。
结论该方法稳定可靠,重现性好,可有效控制酚酞片的质量。
标签:RP-HPLC;酚酞片;含量测定酚酞片为刺激性泻药,口服后在肠内碱性环境中形成可溶性钠盐,刺激结肠而产生导泻作用,作用温和,服药后4~8 h可排除软便[1]。
止泻药及镇吐药,用于治疗习惯性顽固性便秘。
2010年版《中国药典》[2]采用UV-Vis分光光度法对酚酞原料和其片剂测定含量。
本文参阅相关文献[3-8]采用RP-HPLC法测定酚酞片的含量,与UV-Vis分光光度法测得结果一致,RP-HPLC法亦可准确测定出酚酞片中酚酞的含量。
1 仪器与试药高效液相色谱仪(沃特斯Waters 2695),二极管阵列检测器(DAD 检测器);BS 110S(d=0.1mg)、AB135-S(d=0.01 mg)、电子天平(瑞士梅特勒-托利多公司);酚酞对照品(批号:100091-199601,含量为100.0%);酚酞片(批号:1202731、120302、120103);所用试剂均符合色谱条件要求。
2 方法与结果2.1 色谱条件色谱柱:沃特斯Waters C18柱(4.6 mm×150 mm,5 μm),柱温:25℃;流动相:甲醇-0.05 mol/L NH4H2PO4(用磷酸调pH至3.31)(70:30);流速:1 mL/min;检测波长:275 nm;进样量:10 μL。
柱前衍生-反相高效液相色谱法
柱前衍生-反相高效液相色谱法1.引言1.1 概述概述部分的内容可以介绍柱前衍生-反相高效液相色谱法的背景和意义。
下面是一个概述的范例:正如我们所知,液相色谱法是一种常用的分离和检测分析技术,在化学、药学、环境科学等领域具有广泛的应用。
然而,传统的液相色谱法在某些情况下可能面临一些挑战,如分离效果不理想、分析时间较长等。
为了克服这些问题,柱前衍生-反相高效液相色谱法被提出并逐渐受到关注。
柱前衍生是指在样品处理中,在样品中引入适当的衍生试剂,通过与目标分析物发生化学反应,使其在色谱分析中具有更好的分离性能和检测灵敏度。
反相高效液相色谱法是基于分离样品中不同化学性质的分子在反相色谱柱上的亲水作用,达到分离和定量分析的目的。
柱前衍生-反相高效液相色谱法不仅可以提高色谱分析的分离效果,还能够提高检测灵敏度和减少分析时间。
这对于复杂样品的分析具有重要意义,例如药物代谢产物、环境污染物等。
通过引入适当的衍生试剂,可以有效地改善样品的分离性能,同时提高对目标分析物的响应,从而实现快速、灵敏的定量分析。
本文将对柱前衍生-反相高效液相色谱法的原理、方法和应用进行详细介绍。
首先,我们将阐述柱前衍生的基本原理和常用的衍生试剂。
然后,重点介绍反相高效液相色谱法的步骤和关键参数。
最后,我们将通过实例和应用案例来阐述柱前衍生-反相高效液相色谱法在药物分析、环境监测等领域的应用前景。
通过本文的阅读,读者将能够全面了解柱前衍生-反相高效液相色谱法的原理和实践,为他们的研究和实验工作提供参考和指导。
文章结构部分应包括以下内容:本文主要分为三个部分,即引言、正文和结论。
具体结构如下:1. 引言1.1 概述本部分将简要介绍柱前衍生-反相高效液相色谱法的研究背景和意义。
首先,说明柱前衍生技术在分析化学领域的重要性,该技术可以通过将样品与特定试剂反应生成易于分析的化合物,从而提高液相色谱分析的敏感性和选择性。
其次,介绍反相高效液相色谱法在分析化学中的广泛应用,包括药物分析、环境监测和食品安全等领域。
高效液相色谱法测定废水中酚类化合物含量
第19卷·第1期2019年3月0引言随着生活水平的提高,人们对生态环境的关注度越来越高,其中,水体污染已经成为环境污染中最重要的方面[1]。
酚类化合物被广泛应用于染料、医药杀菌剂、防腐剂以及除草剂等各个方面,是我国工业发展不可或缺的一种重要有机化合物[2]。
但是,含酚工业废水对环境危害巨大,具有致癌、致突变以及致畸等严重危害,我国已经把酚类化合物列为5种毒物之一[3,4]。
因此,对酚类化合物的检测十分必要。
目前已有多种检测酚类化合物的方法,例如福林-西莫卡特比色法[5]、分光光度法[6]、气相色谱法[7]以及高效液相色谱法[8]等。
其中,高效液相色谱法具有准确度高、重现性好以及操作简单等优点,因此,本实验采用高效液相色谱法测定废水中间苯二酚和对苯二酚的含量。
1实验部分1.1仪器与试剂LC-20A 高效液相色谱仪(日本岛津公司);FA-1104电子天平(上海越平科学仪器有限公司);KQ5200超声波清洗器(合肥金尼克机械制造有限公司)。
高效液相色谱法测定废水中酚类化合物含量张德谨1,2,王君荷1,谢永1,2,朱军1,李沙沙1,张萍花1(1.宿州学院,安徽宿州234000;2.宿州学院精细化工产品开发研究所,安徽宿州234000)【摘要】采用高效液相色谱法同时测定废水中间苯二酚和对苯二酚的含量,以酚类化合物浓度为横坐标,峰面积为纵坐标,绘制标准曲线,通过精密度试验、稳定性试验以及加标回收率实验对该方法进行评价。
结果表明:测定间苯二酚的线性回归方程为Y=0.00009X+0.31783,测定对苯二酚的线性回归方程为Y=0.0002X-1.36472。
在设定的色谱条件下,模拟废水中间苯二酚浓度为6.27mg/L ,对苯二酚浓度为2.30mg/L ,加标回收率试验、精密度试验和稳定性试验结果表明,该检测方法准确可靠。
【关键词】高效液相色谱法;含量;酚类化合物Determination of Phenolic Compounds in Wastewater by HPLCZHANG De-jin 1,2,WANG Jun-he 1,XIE Yong 1,2,ZHU Jun 1,LI Sha-sha 1,ZHANG Ping-hua 1(1.Suzhou University,Suzhou 234000,China;2.Fine Chemical Product Development Research Institute,Suzhou University,Suzhou 234000,China)【Abstract 】HPLC was employed to determine the contents of resorcinol and hydroquinone simultaneous,the standard curves were used to determine the content of hydroquinone and resorcinol in wastewater,and the method was evaluated by re-covery rate test,precision test and stability test.The results indicated that the linear equation of resorcinol was Y=0.00009X+0.31783and the linear equation of hydroquinone was Y=0.0002X-1.3647.The linear correlation was good.The concentra-tion of resorcinol in wastewater was 6.27mg/L,and the concentration of hydroquinone was 2.30mg/L,the results of recovery rate test,precision test and stability test showed that this method was accurate and reliable.【Key words 】HPLC;content;phenolic compounds 〔中图分类号〕O658〔文献标识码〕A〔文章编号〕1674-3229(2019)01-0059-04[收稿日期]2018-12-11[基金项目]安徽省高校自然科学重点项目(KJ2017A435);安徽省大学生创新创业训练项目(201710379130);宿州学院重点科研项目(2017yzd12)[作者简介]张德谨(1989-),男,在读博士研究生,宿州学院化学化工学院助教,研究方向:绿色化工与可再生资源开发。
反相高效液相色谱法测定
第32卷2004年10月 分析化学(FENXI HUAXU E ) 研究报告Chinese Journal of Analytical Chemistry 第10期1368~1370反相高效液相色谱法测定o 2烯丙基苯酚和p/o 2丁酰基苯酚在番茄中的残留量胡继业1 张文吉2 李建中311(中国科学院生态环境研究中心,北京100085) 2(中国农业大学理学院,北京100094)摘 要 建立了o 2烯丙基苯酚、p/o 2丁酰基苯酚在番茄中残留量的反相高效液相色谱分析方法。
样品前处理过程简便,方法准确可靠,灵敏度和准确度达到了农药残留检测的要求。
o 2烯丙基苯酚、p/o 2丁酰基苯酚在番茄中的最低检测浓度为0.01mg/kg ;方法的添加回收率(0.05~1.00mg/kg )在87.7%~95.1%之间。
关键词 o 2烯丙基苯酚,p/o 2丁酰基苯酚,反相高效液相色谱法,残留分析 2003209212收稿;2004205224接受1 引 言o 2烯丙基苯酚(商品名称绿帝)和p/o 2丁酰基苯酚(商品名称银泰,为p 2丁酰基苯酚和o 2丁酰基苯酚的混合物,在原药中所占比例约为7∶3)是山东省农业仿生应用工程研究中心开发研制的两个新型仿生农用杀菌剂,已获得了国家发明专利和农药临时登记(防治番茄灰霉病),现已批量生产。
它们是以银杏外种皮中的杀菌、抑菌活性化合物的化学结构为模板,经过结构简化,通过人工模拟化学合成得到。
田间药效实验证明,o 2烯丙基苯酚可有效的防治苹果腐烂病、番茄灰霉病、玉米大斑病等病害1;p/o 2丁酰基苯酚对苹果腐烂病、苹果轮纹病、番茄轮纹病、玉米大斑病及小麦纹枯病具有良好的防治效果2,3。
目前这两种农药的残留分析方法,国内外没有文献报道。
作者研究了这两种杀菌剂在番茄中残留的提取、净化方法以及高效液相色谱检测方法,为番茄田中的残留实验、正式注册登记和推广应用奠定基础。
2 实验部分2.1 仪器和试剂Waters 高效液相色谱仪及色谱工作站(美国Waters 公司);RE 252旋转蒸发仪(上海亚荣生化仪器厂);SHB 2Ⅲ循环式多用真空泵(郑州长城科工贸有限公司);20PR 2520高速冷却离心机(日本日立公司);8782B 多功能粉碎机(深圳天南公司);WX 22无油真空泵抽滤器(临海市精工真空设备厂);HZQ 2C空气浴振荡器(哈尔滨东联电子公司);电子天平(Sartorious 公司)。
反相高效液相色谱法测定甘油二酯含量研究
反相高效液相色谱法测定甘油二酯含量研究汪勇;宋坷珂;王丽丽;韩雪;赵谋明【期刊名称】《中国粮油学报》【年(卷),期】2010(025)003【摘要】用分子蒸馏和柱层析的方法制备了大豆油甘油二酯(DAG)的标准样品,通过高效液相-电喷雾质谱联用(HPLC-ESI-MS)和薄层色谱法(TLC)验证了标准样品的纯度.用反相高效液相色谱(RP-HPLC)在UV=210 nm做大豆油DAG的标准曲线,在质量浓度范围1.0~12.0 mg/mL,大豆油DAG标准曲线线性良好.通过加样回收试验表明,加样回收率在104%~109%之间,RSD在0.69%~5.19%之间.并用该方法分析了大豆油、分子蒸馏DAG样品中的DAG.【总页数】5页(P119-123)【作者】汪勇;宋坷珂;王丽丽;韩雪;赵谋明【作者单位】华南理工大学轻工与食品学院,广州,510641;暨南大学食品科学与工程系,广州,510632;暨南大学食品科学与工程系,广州,510632;暨南大学食品科学与工程系,广州,510632;暨南大学食品科学与工程系,广州,510632;华南理工大学轻工与食品学院,广州,510641【正文语种】中文【中图分类】TS227【相关文献】1.柱前衍生化反相高效液相色谱法测定大鼠血浆中牛磺酸的含量及其药动学研究[J], 岳山岚;王维;帅淑平;王静;王吉琳;赵一凡;杨俊毅2.反相高效液相色谱法测定环境有害物质—酚酞含量的研究 [J], 谭丙文;罗志焕;孟庆保;邹振宇3.反相高效液相色谱法测定加味藿香正气丸中橙皮苷含量的研究 [J], 钱方;薛非凡4.反相高效液相色谱法测定加味蕾香正气丸中橙皮苷含量的研究 [J], 钱方;薛非凡;5.反相高效液相色谱法测定益气生血胶囊中黄芪甲苷含量的方法学研究 [J], 孙宾娟因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
反相高效液相色谱法测定环境有害物质—酚酞含量的研究谭丙文 1 罗志焕1(1立创(中山)检测技术服务有限公司,中山,528415摘要(Abstract):本文研究并建立采用反相高效液相色谱法(RP-HPLC-DAD)测定环境有害物质——酚酞含量的方法。
酚酞采用色谱纯甲醇常温超声萃取,色谱分离柱为Agilent Zorbax Eclipse Plus C18分析柱(4.6mm*250mm*5μm),以色谱纯甲醇(100%)为单一流动相等度洗脱测定。
紫外检测波长λ=276nm(定量波长)/229nm(辅助定性波长),TCC柱温箱温度设置为30℃。
标准曲线线性范围1μg/mL~100μg/mL,线性方程A=27.35039C-1.96508*10-2,相关系数0.9999,最低检出限0.37μg/mL,平均回收率101.1%,相对标准偏差RSD为0.06%(n=10)。
该方法简单、灵敏、准确、快速,为我国应对REACH SVHC产品检测及质量控制提供了一个实用便捷的分析方法。
关键词(Key Words): 反相高效液相色谱法(RP-HPLC-DAD) 等度洗脱(Isocratic Elution) 酚酞(Phenolphthalein) 超声萃取(Ultrasonic Extraction)前言(Preface):酚酞,英文学名:Phenolphthalein,分子式C20H14O4,分子量318.33,美国化学文摘社标准CAS号为:77-09-8。
学名:2-[双-(4-羟苯基)甲基]苯甲酸。
无色或微黄色晶体。
熔点257~259℃。
密度 1.277g/cm3。
溶于甲醇、乙醇、乙醚和碱液,难溶于水。
溶于碱液或碱金属的碳酸盐溶液而呈现红色。
在浓碱溶液中则生成无色的三金属盐,红色褪去。
可由邻苯二甲酸酐与苯酚混合后和硫酸共热制得。
化学结构式及标准EI质谱图谱如下图所示:酚酞在实验室中经常用作酸碱滴定的专用指示剂,变色范围pH8.2~10.0,由无色变红。
医药上用作轻泻剂,能刺激肠壁,引起肠的蠕动促进排便,因此目前在一些医用减肥产品或减肥类食品、保健食品常加入酚酞。
已有研究指出:过量服用该类含有酚酞的减肥产品会对人的身体存在极其严重的副作用,建议消费者不要盲目地使用该类产品以保证身体的健康。
而REACH法规则是欧盟规章《化学品注册、评估、许可和限制》(Registration, Evaluation, Authorization and Restriction of Chemicals)的简称,是欧盟于2007年6月1日起就开始实施的化学品监管体系,其目的在于进一步加强对人类健康和环境的保护,保持和提高欧盟化学工业的竞争力。
最近在2011年12月19日,酚酞被REACH主管部门欧盟化学品管理局(ECHA)公布为第六批20种高度关注物质(SVHC)清单中的一种。
当产品中所含此类物质大于0.1%,企业必须向欧盟化学品管理局申请授权或通报,并且必须向采购商和消费者进行公示。
更新至目前REACH法规公布的限制SVHC物质共六批73项。
作为第三方检测机构实验室,开发出快捷、灵敏、准确的分析检测方法以便更好地服务于生产企业,更是刻不容缓。
本文作者开始研发时采用GC-FID(气相色谱-氢火焰离子化电离检测器)及GC-Q-MSD (气相色谱-单四级杆质谱检测器)尝试气相条件下气化并分离酚酞(Phenolphthalein),从而打碎酚酞分子,依次辨别特征碎片离子。
但实验结果反馈的实际情况是:标样浓度点10μg/mL (色谱纯甲醇溶解酚酞标准品并定容),FID色谱峰形很难辨别清晰,MSD做Full-Scan全扫描,无法见到如上图的标准EI质谱图的特征离子碎片,TIC图峰形也难以辨别清楚。
进样100μg/mL也只能勉强辨别一点点色谱峰或质量色谱峰。
正如前文所述,酚酞分子熔点相对较高,气化较难,气相色谱技术应用在酚酞测定上很难开展,于是很自然的想到了应用HPLC技术来分离高沸点热不稳定化合物(这正是高效液相色谱技术的强项!)。
本文作者分析酚酞分子含有两个极性基团—OH,可采用紫外检测器(VWD/MWD/DAD)或ESI-MS/MS (电喷雾离子化-三重串联四级杆质谱检测器)来检测分析酚酞含量。
在综合衡量了成本、效益、灵敏度等因素的“色谱分离三角”后,本文作者主攻选用反相高效液相色谱—光电二极管阵列检测器(RP-HPLC-DAD)来测定酚酞含量,优化色谱分离条件后,方法检出限MDL 满足我司检测要求,且该方法简单、灵敏、准确、快速,为我国应对REACH SVHC产品检测及质量控制提供了一个实用便捷的分析检测方法。
1 实验部分(Experiment Part)1.1仪器、试剂与标准品美国安捷伦公司Agilent 1260 Infinity HPLC,配DAD光电二极管阵列检测器。
KQ5200DB型超声波仪(昆山市超声仪器有限公司制造,40KHZ,功率:200W)。
甲醇(HPLC色谱纯),德国默克公司Merck制造。
酚酞标准品,德国Dr.Ehrenstorfer公司制造,纯度:98.0% ,带标准物质证书。
1.2标准储备溶液的准备根据标物证书纯度标示,准确称取0.1021g酚酞标准品于干净烧杯中,加入10mL色谱纯甲醇,超声助溶,转移至100mL容量瓶中,甲醇润洗烧杯几次转移至100mL容量瓶中,定容至刻度,即成1000μg/mL标准储备液,依次稀释成1μg/mL、2μg/mL、5μg/mL、10μg/mL、20μg/mL、50μg/mL、100μg/mL系列标准曲线使用溶液。
本文采用10μg/mL浓度点进行方法开发的源头。
1.3样品前处理取代表性的样品,剪碎至2mm×2mm,混匀。
称取样品1.0g(精确至0.001g)样品,置于50mL具塞试管中,加入10mL甲醇溶液,密封好塞子,在超声清洗器中室温提取60min。
然后过0.45μm过滤头过滤至2mL样品瓶中,上HPLC测试。
1.4高效液相色谱条件色谱分离柱:Agilent Zorbax Eclipse Plus C18分析柱(4.6mm*250mm*5μm);流动相:甲醇(100%)为单一流动相等度洗脱,流动相流速:1 mL/min;紫外检测波长λ=276nm(定量波长)/229nm(辅助定性波长),带宽:4nm,狭缝:2nm;TCC柱温箱温度:30℃;进样量:20μL;DAD光谱扫描波长λ:190nm~400 nm,步进值:1 nm。
2 结果与讨论(Analytical Results & Discussion)2.1流动相及色谱分离条件的优化选择据查文献,多数文献报导推荐使用甲醇/水流动相体系,比例有的60%:40%,有的70%:30%,有的添加了1%磷酸(H3PO4)缓冲盐,有的添加了0.5%冰醋酸(HAc),如此等等。
本文作者方法开发思路为:在力求保证分离度、灵敏度、分析速度的“色谱分离三度”的大前提下,尽量简化流动相的配比组分,因此项目组分较为单一简单,先不采用缓冲盐,而采用甲醇/水流动相体系进行分离,若调试后仍不能满足“三度”,则再配制缓冲盐进行优化调试。
另外可通过DAD紫外光谱扫描功能,确定酚酞(Phenolphthalein)的最佳吸收波长λ。
方法开发第一针的摸索条件:甲醇:水=60%:40%,流动相流速:1mL/min,紫外检测波长λ=?未知,只能在紫外区间任意设置一个。
得出的第一张HPLC谱图如下图:初步确定该图示的色谱峰为酚酞(Phenolphthalein),打上标记,查阅该色谱峰顶的3D光谱图为下图:通过查阅酚酞(Phenolphthalein)紫外吸收光谱图,确定该色谱峰为目标化合物色谱峰,光谱图完全匹配。
从光谱图确定229nm为第一特征灵敏度吸收峰,276nm为第二特征灵敏度吸收峰。
查看两条谱线的HPLC色谱图比较得出,虽然λ1=229nm灵敏度是λ2=276nm灵敏度的将近6倍,但λ1=229nm HPLC色谱图明显杂质峰较多,λ2=276nm HPLC色谱图杂质峰较少,干扰较少,故选择λ2=276nm谱线作为定量波长,选择λ1=229nm谱线作为辅助定性波长。
从而确定最佳吸收波长。
接下来的事情就是优化HPLC色谱峰形,让色谱峰更加“尖锐点”,从而更好地降低信号检出限、方法检出限。
从第一张HPLC色谱图来看,目标化合物峰形出现前伸峰,且10ppm 信号也不是很高。
经验告诉我们:很明显就是甲醇/水流动相系统某一组分含量不够导致HPLC色谱峰形助推动力不足,从而不够“尖锐”!这样无助于降低信号检出限、方法检出限,调高水相比例如何?甲醇:水=50%:50%,进样调试,得出的HPLC谱图如下图:信号降低了,通过查阅该图的峰顶点3D光谱图并比较上图,得出该色谱峰并不是酚酞(Phenolphthalein),另外通过On-line Plot在线谱图窗口观察:在设定的分析时间内,目标化合物Phenolphthalein没有出峰,而是在大约12.30min出峰。
通过第二针的谱图我们可以得出以下结论:流动相体系甲醇组分含量需要“更猛一点”,甚至直接来个100% ,从而将色谱峰形推动得更加“尖锐”,信号更高,无扁头峰、前伸峰、拖尾峰,信号检出限、方法检出限自然能降得更低。
但一个要注意的前提是:不能将目标峰助推到和杂质峰重合在一起。
方法开发的中间步骤可以多设几步流动相组分依次改变的细化条件来实现逐步优化,现在的方向是:依次调高甲醇(Methanol)组分,比较得出最佳色谱峰形。
甲醇:水=90%:10%,得出的色谱图为下图:色谱峰形变得比较尖锐了,信号也提高了,但仍出现前伸峰。
改为甲醇:水=100%:0%,得出的色谱图为下图:流动相系统甲醇比例已经是100%了,峰形尖锐,峰宽狭窄,呈现出较为对称的高斯峰形(Gaussian Peak),同时信号已经提高到最高,再通过点击峰顶点查看3D光谱图,确定该HPLC色谱峰为酚酞(Phenolphthalein),无杂质干扰。
从而确定最优化流动相比例为:甲醇(Methanol)=100%,等度洗脱,4min内完成色谱分离,同时通过3D-光谱图来进一步辅助定性判断样品是阳性(Positive)抑还是阴性(Negative)。
通过最优化的HPLC色谱图,初步判断最低浓度点能做到1μg/mL,下面将作标准曲线线性范围的讨论。
2.2标准曲线线性实验精密称取0.1021g酚酞标准品于干净50mL烧杯中,加入10mL色谱纯甲醇,超声助溶,转移至100mL容量瓶中,甲醇润洗烧杯几次转移至100mL容量瓶中,定容至刻度,即成1000μg/mL标准储备液,依次稀释成1μg/mL、2μg/mL、5μg/mL、10μg/mL、20μg/mL、50μg/mL、100μg/mL系列标准曲线使用溶液。