GSK3β_β-catenin signaling is correlated with the differentiation of glioma cells induced by wogoni
β-catenin在子宫内膜癌组织中的研究

β-catenin在子宫内膜癌组织中的研究孔翠花;徐韶杰【摘要】目的探讨β-catenin在子宫内膜癌组织中的表达情况及其与子宫内膜癌发展及转移的关系.方法选择子宫内膜癌手术患者的手术切除标本80例及正常子宫内膜手术切除标本20例,采用免疫组织化学S-P法检测β-catenin 的表达,分析其在组织中表达的差异及其与临床病理特征的关系.结果在子宫内膜癌组织中β-catenin蛋白的阳性表达率明显高于正常子宫内膜组织(P<0.05),在不同组织病理分级中的表达结果 :G1级的阳性率为86.9%,G2级的阳性表达率为80.6%,G3级的阳性表达率为42.3%,三者之间差异有统计学意义(P<0.05);子宫肌层浸润<1/2病例的阳性率为56.5%,肌层浸润≥1/2者阳性率为87.3%,两者差异有统计学意义(P<0.05);淋巴结转移阴性的病例中的阳性率为59.3%,淋巴结转移阳性者为84.6%,两者之间差异有统计学意义(P<0.05);手术病理分期Ⅰ期的阳性率为63.6%,Ⅱ期的阳性率为77.3%;Ⅲ期的阳性率为80.0%,Ⅰ期与Ⅱ期差异有统计学意义(P<0.05),Ⅱ期与Ⅲ期差异无统计学意义(P>0.05).结论β-catenin在子宫内膜癌组织中呈高表达,且与组织分级、子宫肌层浸润及淋巴结转移有关,表明β-catenin在子宫内膜癌的发生、发展、侵袭及转移中发挥作用.【期刊名称】《河北医药》【年(卷),期】2010(032)017【总页数】3页(P2320-2322)【关键词】子宫内膜癌;β-catenin;免疫组化【作者】孔翠花;徐韶杰【作者单位】054000,河北省邢台市,邢台医学高等专科学校第二附属医院妇产科;解放军第316医院妇产科【正文语种】中文【中图分类】R737.33子宫内膜癌是最常见的女性生殖道恶性肿瘤,占女性生殖道恶性肿瘤的 20%~30%,而发生率逐年增高[1]。
本研究旨在通过免疫细胞化学检测子宫内膜癌组织中β-catenin的表达情况,分析其在子宫内膜癌组织中的表达与子宫内膜癌临床病理特征的关系,为子宫内膜癌预后的判断、治疗方案的选择提供参考。
上皮细胞转化大肠发展

上皮细胞转化大肠发展当前,大肠癌已是欧洲排名第2位的肿瘤致死性疾病,占肿瘤致死性疾病的10%左右.引起大肠癌发生的原因众多,如基因突变引起的家族性腺瘤性息肉病(familialadenomatouspolyposis,FAP)、DNA错配修复基因(MMR)突变引起的遗传性非息肉性结肠癌(herediarynonpolyposiscol-orectalcancer,HNPCC)、Crohn''''s病、溃疡性结炎、上皮细胞内癌基因激活和抑癌基因失活等均与大肠癌的发生发展密切相关1.上皮细胞间质转化(epithelial-mesenchymaltransition,EMT)是指在生理或病理情况下发生细胞上皮-间质转变,同时伴随细胞形态与相关基因表达的改变.EMT在胚胎形成以及组织器官发育过程中也起着重要作用(如中胚层和神经冠结构形成以及心脏形态发生过程2),但也可引起器官纤维化和参与肿瘤形成过程,在此过程中上皮细胞顶-底极性改变、桥粒等紧密连接结构消失、细胞骨架重组,波形蛋白表达上调、角蛋白表达下调,从而使细胞离体、获得迁移水平,并能抵抗细胞凋亡3.近来研究发现EMT在大肠癌发生发展过程中起着重要作用,并与肿瘤细胞的浸润和转移有着非常密切的关系.1EMT在炎症促动大肠癌发生发展中的作用慢性炎症被认为是包括大肠癌在内的很多种人类肿瘤疾病的原因之一,流行病学和临床研究均证实两种主要的炎症性肠病(inflammatoryboweldisease,IBD):溃疡性结肠炎和Crohn''''s病4发展成大肠癌的风险明显升高.慢性炎症能够通过诱导细胞DNA修饰导致肠上皮细胞发育不良,此外还可通过DNA甲基化和组蛋白修饰等作用影响肠上皮发育过程5.Bataille等6在Crohn''''s病瘘管形成过程中发现肠道上皮标志物E-cadherin和?-catenin表达降低(?-catenin 在EMT的起始阶段合成增加,而在EMT最终阶段合成明显减少7);间质标志物?-integrin表达增多(TGF-?激活EMT的过程依赖于?-integrin,然后通过Smad3依赖性的转录过程或者非Smad3依赖性、p38MAPK激活和GTP酶调节的信号传导途径8),而该蛋白随着EMT的进展逐渐由胞膜向胞质、胞核转移,?-catenin移位被认为是Crohn''''s病发展过程中EMT的关键分子步骤.在肿瘤相关炎症(cancerrelatedinflammation,CRI)相关分子中发现一些重要的始动因子,包括NF-?B、STAT39、IL-1?、IL-6、IL-10和TNF-?等.NF-?B是一种关键的内源性炎症/免疫调节因子10.Douglas等在大肠癌细胞中发现了异常的NF-?B调节,且证实在结肠肿瘤起始和发展中NF-?B和CRI之间存有密切联系,通过靶向灭活IkappaB使肿瘤浸润白细胞中NF-?B失活抑制了炎症相关大肠癌的发生,从而为结肠肿瘤中NF-?B和炎症细胞的作用提供了基因水平证据.IL-6是NF-?B激活的一个主要效应分子,并且与STAT3存有密切关系,他具有促动生长和抗凋亡水平11.研究发现IL-6能保护正常肠上皮细胞和癌前细胞免受凋亡,并促动肿瘤起始细胞增殖,在此过程中NF-?B-IL-6-STAT3通路起着重要作用.Lee等12发现大肠癌中NF-?B的活化状态需要STAT3维持,提示STAT3是肿瘤细胞增殖和存活的关键因子,并调控了c-Myc、Mcl-1、CyclinD和Bcl-2表达13.抑制因子从不同水平上调控NF-?B信号通路,Tir8是表达于肠黏膜上IL-1R家族的一员,他能够通过阻止IRAK-1和TRAF-6,抑制信号从IL-1R/TLR复合物传导14.在小鼠大肠癌肿瘤模型中发现NF-?B下游分子CCL2、CCL3、IL-1和IL-6能够促动炎症相关的肿瘤形成,并发现NF-?B激活过程中Tir8的缺失直接导致了大肠癌形成15.肿瘤相关巨噬细胞(tumorassociatedmacrophages,TAM)分泌的TNF通过抑制GSK-3?促动了Wnt/?-catenin信号传导,促动了结肠上皮细胞向间质转化,此过程在大肠癌发展中起着重要作用16.此外,炎症细胞中NF-?B激活也造成了COX-2和ROS水平升高,ROS能诱导DNA损伤、DNA甲基化、转录后修饰和肿瘤抑制基因突变等7;控制炎症反应和诱导肿瘤细胞凋亡的TGF-?和低氧诱导因子-1(hypoxia-induciblefac-tor-1,HIF-1)同样是炎症微环境中促使上皮细胞发生间质转化的潜在诱导因子17.Grivennikov等17证实IL-6与其受体sIL-6?结合后停留在细胞表面并能借助胞内TGF-?通路促动结肠上皮向恶性转化;IL-10激活STAT3(信号传导蛋白-转录激活物)后通过与IL-6相似的途径介导细胞恶性转变18.TGF-?作为炎症因子可造成包括肠道在内的多器官自身免疫性疾病,且可激活多种信号通路如Erk、c-Jun、JNK、PI3K和RhoA等19,也能诱导某些转录因子和转录调节因子在EMT 中的表达,包括?EF1、SIP1和Snail等,从而有利于结肠上皮EMT的发生20.TNF-?在IBD发病机制中是一种重要的炎症因子,在炎症相关大肠癌(colitisas-sociatedcolorectalcancer,CAC)中也起着重要作用21,TNF-?在CAC中主要依赖激活胞内转录因子NF-?B实行信号传导,通过NF-?B的多向性转录激活作用(NF-?B能够结合至靶基因MMP-9、IL-8、uPA、VEGF、CXCR4、骨桥蛋白等的启动子或增强子之上实行调控22)诱导结肠上皮细胞向肿瘤细胞分化、增殖,并抑制细胞凋亡、促动肿瘤侵袭和转移23.此外,其他炎症因子如IL-12、IL-13和INF-?等在慢性大肠炎发展过程中也参与了肿瘤形成过程,而TGF-?、IL-10则能在此过程中发挥协同作用24.当前已证实上述参与炎症发生和发展的各种细胞因子如,TGF-?、TNF-?和NF-?B等均是EMT信号通路的关键因子,可见EMT参与了炎症促动大肠癌发生和发展的相关过程,但是EMT在此过程中的详尽机制尚待进一步研究.2EMT在腺瘤性息肉病相关的大肠癌发生发展中的作用FAP在结肠腺瘤性息肉疾病中占有主要地位,相关研究证实位于染色体5q21的APC突变失活是FAP的主要原因25,APC突变被认为启动了大肠癌发生的多步骤过程,最终FAP往往发展成为大肠癌26.与FAP相同的是绝绝大多数散发大肠癌病例起源于结肠腺瘤且同时伴有APC突变.Vécsey-Semjén等27证实小鼠敲除APC外显子exon14后可导致结肠腺瘤发生,免疫组织化学检测该模型结肠上皮细胞中可见Wnt信号通路的关键因子?-catenin在胞质和胞核中积累,且编码C-Myc和CyclinD1的mRNA也显著增加.APC是一种肿瘤抑制基因,能够作为Wnt/?-catenin的负性调控因子,在正常结肠上皮细胞APC/?-catenin复合物被丝氨酸-苏氨酸激酶(GSK3?)磷酸化,导致?-catenin降解,而在APC突变失活及Wnt信号转导通路开启时GSK3?的磷酸化作用被抑制28,使其不能诱发?-catenin降解,从而造成胞质内的?-catenin持续累积,后者作用于靶基因C-Myc和CyclinD1等,最终导致Wnt通路介导的EMT 发生,正常结肠黏膜上皮向间质转化,最终结肠上皮细胞发生恶性转化29,30.Lochter等31利用COGA-8结肠上皮细胞培养发现CyclinD1与CDK4、CDK6结合,诱导生成CyclinA和CyclinE,再与CDK2结合从而使结肠上皮细胞从G1期进入S期.C-Myc启动子区域有?-catenin结合位点,所以C-Myc表达能够被Wnt通路上调,C-Myc过表达使其结合至CyclinD2启动子特定的DNA序列并促动CyclinD2的转录过程32.Wnt通路还能直接上调CyclinD1,因为CyclinD1启动子区域包含LEF-1结合位点,而该位点被认为是Wnt通路的直接作用靶点33.?-catenin在胞核内与淋巴细胞增强子结合因子1(LEF-1)和T细胞因子-4(Tcf-4)结合并作为转录共激活子启动下游基因(Slug、Cdx-1、Id2和ENC1等)表达.APC上?-catenin结合位点的减少水准与Wnt通路中?-catenin/LEF-1/Tcf-4复合物增加的水准呈负相关34,而?-catenin/LEF-1/Tcf-4的增加导致了结肠上皮细胞间紧密连接蛋白ZO-1减少、胞间桥粒等连接蛋白降解、细胞骨架重组、细胞离体和获得迁移水平35,还能够直接作用于AP-1转录因子复合物中c-jun和fra-1的启动子部位使该转录复合物增多、上调uPAR转录36.此外,该复合物还能上调ZEB1表达(高表达于FAP腺瘤、结肠腺癌上皮细胞,且与胞核?-catenin水平呈正相关37),而ZEB1能抑制E-cadherin生成38,且该转录激活复合物参与了腺瘤转变为腺癌甚至肿瘤转移的全过程39.以上过程最终使结肠上皮细胞经历EMT过程(如细胞间连接蛋白降解、细胞迁移水平增强和获得间质表型等)并向恶性转化.另外,CK2?是一种高度保守的丝氨酸-苏氨酸蛋白激酶,能够磷酸化多种底物并在多种生理病理过程中起重要作用40.正常结肠上皮细胞逐渐演变成腺瘤或腺癌的过程被认为与EMT及E-cadherin、Vimentin和?-catenin等基因表达改变紧密相关.Zou等40发现CK2?表达于正常结直肠上皮细胞和结直肠腺瘤/腺癌细胞,通过调节参与细胞周期的癌基因c-myc和抑癌基因p53和p21等影响了大肠癌的演变过程.CK2?敲除或转染CK2?SiRNA后上皮标志物E-cadherin表达显著升高、间质标志物vimentin表达降低,还能造成细胞中转录因子Snail1、Smad2/3和癌基因c-myc的表达下降,以上结果说明CK2?能够对上皮间质转变起到某种水准的抑制作用,但是CK2?影响结肠腺瘤向大肠癌转变的具体机制尚未完全明确.3microRNAs介导的EMT在大肠癌发生发展中的作用microRNAs是一类长度在18-25nt的单链寡核糖核苷酸的非编码RNA,具有高度的保守性、组织特异性和发育时序性41,在转录后水平通过负向调节mRNA发挥其功能,与mRNA的靶向识别以与3''''末端UTR互补性结合为基础42.mi-croRNAs翻译水平的抑制作用常伴随由poly(A)尾加速脱腺苷化和后续核酸外切消化导致的靶mRNA水平减少43,而且microRNAs控制其靶点特异性的关键区域在5''''末端2-7个碱基对的种子序列44,能够在细胞增殖、分化、凋亡、新陈代谢及胚胎发育等过程中起调控作用45,部分microRNAs通过调控癌基因和肿瘤抑制基因的表达;部分通过直接作为癌基因或肿瘤抑制基因参与了大肠癌的病理过程46.虽然microRNAs参与大肠癌发生发展的相关研究较多,但相关microRNAs在大肠癌中介导EMT的研究仍较少.研究证实在很多原发肿瘤及相对应转移瘤中存有不同水准的microRNA表达,在蛋白络氨酸磷酸酶(PTP)Pez诱导MDCK的细胞系中发现了TGF-?参与EMT的过程,因为在该细胞系中发现了细胞间连接缺失和间质表型过表达.此外通过RT-PCR还发现miR-200家族(miR-200a、miR-200b、miR-200c、miR-141和249)及miR-205表达的下调,而稳定的miR-200s过表达能够阻止TGF-?诱导的EMT,提示miR-200s是EMT的关键调控因子,且miR-200s是通过抑制ZEB1和SIP1的翻译来调控EMT的47.关于miR-200家族、ZEB1和SIP1,我们推测上皮细胞和间质细胞表型之间的转化是由ZEB1和SIP1的水平决定的.ZEB1和SIP1结合至目的基因如上皮细胞关键基因E-cadherin启动子成对的ZEB样E-Boxs(CACCTG)上从而抑制这些基因的转录48.以上证据提示miR-200s的丢失可能导致肿瘤的侵袭性增强,甚至造成远处转移.TGF?、TNF?由浸润的炎症细胞或肿瘤细胞产生,已被证实能够诱导结肠癌细胞发生EMT,在结肠癌SW480细胞系中通过miRNA表达分析发现稳定敲除ZEB1能够导致胞间连接蛋白E-cadherin表达上调、细胞迁移和侵袭的水平下降,而miR-141、miR-200b和miR-200c的表达水平明显升高,且miR-141、miR-200c的表达上调最为明显.此结果也被RT-PCR检测所证实,提示这两种miRNA参与了结肠癌EMT过程49.而Colwell50利用TGF?/TNF?诱导LIM1863大肠癌细胞发生EMT的过程中发现miR-21和miR-31表达水平明显升高;蛋白定量分析发现miR-21和miR-31促动了TGF?诱导的EMT的过程,与miR-200抑制EMT上游调控因子ZEB1/2不同,miR-21和miR-31主要作用于EMT下游因子T淋巴瘤侵袭转移蛋白1(TIAM1),后者是一种RacGTPase交换因子51.此外,miR-9和miR-335通过直接抑制E-cadherin和SOX4合成促动了大肠癌细胞转移.以上结果说明某些microRNA能够在TGF-?信号通路的下游发挥作用从而促动结直肠癌的发生和转移.启动子超甲基化和肿瘤抑制基因沉默是肿瘤形成的重要分子标志.Davalos等52通过大肠癌原发肿瘤微切除证实5''''-CpG岛超甲基化相关的miR-200b/200a/429和miR-200c/141多顺反子转录沉默是调节EMT和MET转变的重要步骤,也是大肠癌肿瘤进展的关键,并发现miR-200超甲基化和ZEB1/ZEB2上调与CDH1、CRB3和LGL2表达下调相关;TGF?诱导的EMT中miR-200超甲基化失活伴随着E-cadherin丢失和Vimentin增加53;Twist基因启动子获得CpG岛超甲基化后即可诱导结肠上皮细胞发生间质转化.此外,其他组蛋白修饰基因,如LSD1、CREB结合蛋白、SIRT1等也参与了EMT过程.deKrijger等46发现36%的大肠癌原发肿瘤中miR-34a表达下降,部分归因于TP53的突变,部分是因为启动子甲基化;EMT激活因子TGF?上调也促动了miR-21和miR-31表达,后两者在大肠癌中促动了TGF?诱导的EMT过程.此外,miR-373、miR-126和miR-196a转染的大肠癌细胞则显示出明显肺转移潜能.Sreekumar等54证实E-cadherin表达受miR-9直接调控而转录抑制,miR-199和miR-218则是间质特征性蛋白N-cadherin的潜在直接调控mi-croRNA.miR-138、miR-488和miR-151能够在EMT过程中调节FAK的表达水平从而影响大肠癌肿瘤细胞的迁移水平.4大肠癌中EMT的相关信号通路在EMT介导大肠癌发生发展过程中伴随着众多信号通路的激活,将胞外信号传导入胞内引起E-cadherin、Vimentin等异常、表型改变、基底膜降解、上皮细胞向间质转变和细胞迁移等一系列变化,最终导致正常结肠上皮细胞转变为大肠癌细胞.4.1Wnt/?-catenin和FGF信号传导通路Wnt信号需要通过标准和非标准的信号通路传导,FGF信号通过PI3K-AKT、MAPK和PLC?通路传导.GSK3?是Wnt标准信号传导通路和FGF依赖性PI3K-AKT信号传导通路的关键分子.标准的Wnt信号通路决定细胞的分化方向,非标准的信号通路控制细胞的极性和运动潜能,前者通过卷曲家族受体(Frizzled)和LRP5/LRP6受体实行信号转导,后者通过Frizzled和ROR1/2共同受体实行信号传导.LRP5和LRP6是LDL家族的蛋白分子,胞外有Wnt结合位点,胞内有Axin模体结构.除Wnt信号外,?-catenin为实现在蛋白化及蛋白酶体介导的降解而与GSK3?结合并被后者磷酸化55,标准的Wnt信号诱导Friz-zled-Dishevelled复合物与LRP5/LRP6-AXIN-FRAT结合,使?-catenin从GSK3?释放,最终在胞核中稳定的累积.?-catenin与TCF/LEF、BCL9/9L结合成TCF/LEF-?-catenin-Legless-PYGO复合物,作为Wnt信号通路的效应物56.非标准Wnt信号通路中ROR1和ROR2是受体型络氨酸激酶,胞外为Wnt结合位点,胞内为CKI?结合结构域,RhoA、c-JUN、N末端激酶(JNK)、Nemo样激酶(NLK)是非标准Wnt信号通路的效应物.FGF与受体结合后诱导受体二聚化、络氨酸激酶活化及受体自身磷酸化,FRS2、FRS3和PLC?与磷酸络氨酸残基相互作用被募集至FGF 受体,然后FRS2/3招募GRB2、SHP2,FRS2/SHP2/GRB2复合物募集GAB1激活PI3K,导致GSK3?活性下降、Snail-EMT级联反应激活、E-cadherin/?-catenin表达下调、结肠上皮细胞恶性转化,最终参与了大肠癌的发生发展过程57.基因分析发现Dishevelled是Wnt通路的正向调控蛋白,它发挥作用时处于受体下游、?-catenin的上游,能导致c-Myc和cyclinD基因在结肠癌细胞中出现扩增58.4.2Ras信号传导通路Ras蛋白在调节大肠癌发生发展的信号通路中起着重要作用.生长因子活化的受体激活后,活化的Ras通过Raf激酶激活MAPK激酶(MEK1、MEK2),导致胞外ERK1、ERK2激活59.ERKs位移至细胞核并激活核转录因子,如Elk-1、ATF-2、ETS1/2,使癌基因转录迅速开启.首先,BRAF是Raf家族的一员,他的突变与增强了的激酶活性相关,且在9%-11%的结直肠癌中发现此现象;其次,在30%-40%的腺瘤和76%的结直肠癌肿瘤中证实MEK磷酸化及激活;再次,结直肠癌中表现ERK的高度激活,且证实ERK1、ERK2活性在肠道肿瘤中明显升高;最后,试验证实阻断MEK/ERK抑制了结肠癌细胞的生长,表明ERK参与了结肠癌细胞的增殖.MEK1通过Egr-1、Fra-1增强Snail1/2表达而下调E-cadherin60.另有研究证实结肠上皮细胞表达活化的MEK1获得了向肿瘤细胞转变及转移的潜质.除MAPK途径外,Ras还可通过PI3K和RhoGTPase或与TGF-?协同诱导大肠癌EMT.PI3K激活后影响EMT过程的机制如前所述;此外,PI3K/Akt能够使RhoGTPase激活,也能与TGF?通路相互作用从而影响大肠癌EMT过程61.4.3PI3K信号传导通路磷酸酰肌醇3激酶(PI3K)通路对正常细胞代谢、生长及肿瘤进展起着重要作用.PI3K的抑制因子PTEN缺失(10号染色体上磷酸酶和张力蛋白同源敲除)与结肠肿瘤相关,证实PI3K信号通路在大肠癌发生过程中起着重要作用.近年来研究提示66%-70%的大肠癌中PTEN表达下调,且与微卫星不稳定紧密相关.此外还证实TNF?造成了PTEN下调.IEC-6和HIE细胞敲除PTEN后?-catenin表达水平明显升高,大肠癌SW480细胞中V-catenin表达也有轻度升高,提示?-catenin的表达受PTEN调控.利用LY294002或Wortmannin抑制PI3K降低了c-Myc和cyclinD1表达,而在RIE-1细胞中敲除PTEN则显著上调了上述蛋白的表达;同样在IEC-6和HIE细胞中使PTEN沉默增加了c-Myc和cyclinD1的表达,说明PTEN丢失能够通过促动细胞增殖、抑制凋亡而影响大肠癌的发生62.Bowen等63发现在野生型PTEN结肠肿瘤细胞中Akt2过表达导致了肿瘤微转移的形成,提示Akt可能是作为TGF-?1下游调控因子发挥作用的.PI3K/Akt通路下游的效应分子mTOR激酶调控着CRC肿瘤的发生,且证实mTORC1和mTORC2通过RhoA和Rac1通路调控着结肠癌细胞的迁移水平.4.4Notch和Hedgehog信号传导通路Notch通路在胚胎发育和内环境稳定中发挥着重要作用,其异常激活参与了多种肿瘤的发展过程.该通路激活是由Notch配体(Jag1、Jag2、DLL1、DLL3和DLL4)结合至相对应受体上引起的.配体在细胞外被蛋白酶裂解、在胞内被?-分泌酶裂解64,使得Notch胞内结构域(NICD)转至细胞核,在胞核中与CSL和其他共刺激因子如Maml1、2、3构成转录激活复合物,最终激活Notch通路的靶基因Hes和Hey1,使上皮细胞恶性转化.研究显示大肠癌EMT的诱导因子ZEB1可通过抑制miR-200表达而稳定Notch通路的Jag1、Maml2和Maml3,并证实ZEB1通过提升Jag1的表达而增强Notch通路激活通路存有交互作用66.虽有研究67证实Hedgehog通路能诱导Snaill表达上调,但是尚未证实Hedgehog对Snaill有直接转录激活作用.另外Hedgehog能诱导JAG2表达上调、促动TGF?分泌68,TGF-?1激活使胞核内NF-?B调控的ZEB1和ZEB2表达上调,同样使Smad-Sp1调控的间质标志物Vimentin等表达上调从而促动大肠癌细胞迁移及侵袭.5结论在胚胎发育中EMT是必需的生理过程,而肿瘤组织诱导产生的EMT则是肿瘤浸润和转移的重要机制之一.当前,越来越多的证据表明EMT在与肿瘤发生、发展机制中起着重要的作用,随着对EMT作用机制的研究持续深入以及对其信号通路和关键分子的逐步了解,相关的研究结果为临床治疗肿瘤提供了重要的靶点与途径.因为EMT主要是由E-cadherin 的转录抑制因子诱发的,所以借助靶向抑制Snail等的治疗方法为防止肿瘤进展提供了可能.此外,EMT信号传导通路中的关键分子GSK-3?、PAK和TGF?等将来也有可能成为阻断EMT的重要靶点.虽然当前对EMT 发生机制的研究尚未完全清楚,但是随着相关研究的持续深入,人们对EMT的了解将会变得更加清晰.上皮细胞转化大肠发展。
SMK-17在β-连环蛋白突变的癌症中选择性诱导凋亡

SMK-17,a MEK1/2-specific inhibitor,selectively induces apoptosis in b -catenin-mutated tumorsMasaki Kiga 1,2*,Ayako Nakayama 1*,Yuki Shikata 1,Yukiko Sasazawa 1,Ryo Murakami 2,Toshiyuki Nakanishi 2,Etsu Tashiro 1&Masaya Imoto 11Department of Biosciences and Informatics,Faculty of Science and Technology,Keio University,Yokohama,Japan,2Shinagawa R&D Center,Daiichi Sankyo Co.Ltd,Tokyo,Japan.Although clinical studies have evaluated several MEK1/2inhibitors,it is unlikely that MEK1/2inhibitors will be studied clinically.BRAF mutations have been proposed as a responder marker of MEK1/2inhibitors in a preclinical study.However,current clinical approaches focusing on BRAF mutations have shown only moderate sensitivity of MEK1/2inhibitors.This has led to insufficient support for their promoted clinical adoption.Further characterization of tumors sensitive to MEK inhibitors holds great promise foroptimizing drug therapy for patients with these tumors.Here,we report that b -catenin mutations accelerate apoptosis induced by MEK1/2inhibitor.SMK-17,a selective MEK1/2inhibitor,induced apoptosis in tumor cell lines harboring b -catenin mutations at its effective concentration.To confirm that b -catenin mutations and mutant b -catenin-mediated TCF7L2(also known as TCF4)transcriptional activity is a predictive marker of MEK inhibitors,we evaluated the effects of dominant-negative TCF7L2and of active,mutated b -catenin on apoptosis induced by MEK inhibitor.Indeed,dominant-negative TCF7L2reduced apoptosis induced by MEK inhibitor,whereas active,mutated b -catenin accelerated it.Our findings show that b -catenin mutations are an important responder biomarker for MEK1/2inhibitors.Constant activation of the mitogen-activated protein kinase (MAPK)pathway due to aberrant activation of receptor tyrosine kinase and due to K-Ras mutations or BRAF mutations is common in human tumors and represents a major factor in abnormal cell growth 1.Approximately 30%of all human tumors contain an activating Ras mutation 2.Oncogenic V600E mutations in BRAF have been found in 66%of melanomas and in 69%of papillary thyroid tumors 3,4.Furthermore,aberrant activation of the MAPK pathway correlates with tumor progression and poor prognosis in patients with various tumors.The constitutive expression of MEK1/2is sufficient to induce transformation 5,6.Targeting MEK1/2with small-molecule inhibitors is an attractive treat-ment strategy,as all potentially aberrant oncogenic signaling upstream is preventable 7.Furthermore,several MEK inhibitors (e.g.,PD184352/CI-1040and PD0325901)have been evaluated in clinical studies 8–10.However,MEK inhibitors have met with limited clinical success in single-agent therapy.Wnt signaling also plays a central role in cell proliferation and differentiation 11.In the absence of a Wnt stimulus,b -catenin interacts with AXIN1/2,glycogen synthase kinase-3b (GSK-3b ,encoded by GSK3B),and the adenomatous polyposis coli protein (APC).GSK-3b phosphorylates b -catenin and triggers its ubiquitination and degradation by b -Trcp 12.Activation of the Wnt pathway inhibits GSK-3b -dependent phosphorylation of b -catenin and then stabilizes b -catenin.The form of b -catenin resulting from hypophosphorylation then translo-cates to the nucleus and interacts with TCF7L2,leading to increased expression of c-Myc or cyclin D113,14.Mutations in b -catenin enhance its stability and promote the subsequent transactivation of TCF7L2;such transactivation is found in a wide variety of human tumors 15.Although MAPK and Wnt signals are important intracellular signaling pathways,the mechanism of their crosstalk is not yet fully elucidated.In this study,we classified human tumor cell lines as either sensitive or resistant to a MEK inhibitor,as determined by apoptosis induction.We show that mutated b -catenin in tumor cells promotes MEK inhibitor-induced apoptosis.Our results suggest that b -catenin mutations are a novel predictive marker of MEK inhibitors.OPENSUBJECT AREAS:CHEMOTHERAPYAPOPTOSISReceived24September 2014Accepted24December 2014Published2February 2015Correspondence and requests for materials should be addressed to M.I.(imoto@bio.keio.ac.jp)*These authors contributed equally tothis work.ResultsSMK-17inhibited cell proliferation in tumor cell lines with activated K-Ras or BRAF mutations .SMK-17was a potent and highly selective MEK1/2inhibitor with an IC 50of 62and 56nM,respectively (Figure 1A).Several studies have reported a wide range of sensitivity toward the anti-proliferative effects of MEK1/2inhibitors 16.As we have previously confirmed,MEK1/2inhibition by SMK-17without off-targeting kinases has remarkably high selectivity 17;thus,we examined the effect of SMK-17on several types of human tumor cell lines.As shown in Figure 1B,cell lines with BRAF mutations,including colo-205,SK-MEL-1,HT-29,colo-201,and A375cells,were sensitive to SMK-17.Cell lines with K-Ras mutations,such as SW480,HCT 116,SW620,LS-174T,and OVCAR-5cells,were moderately sensitive to SMK-17.Scatter plots showing the log IC 50of cell lines with mutations in the MAPK pathway,including mutations in K-Ras or BRAF,revealed that these cell lines were completely sensitive to SMK-17(Figure 1C).We similarly analyzed the effect of SMK-17in cells with mutations in the PI3K pathway (including mutations in PI3K or PTEN),p53,and the Wnt pathway including APC and b -catenin.Significant differences were not observed in cell lines harboring PI3K and p53mutations.On the other hand,there were significant differences in sensitivities between cell lines with Wnt pathway mutations and theother cell lines (Figure 1C).Similar results were obtained when another MEK inhibitor,U0126,was used instead of SMK-17(Figure 1D).SMK-17induced apoptosis in active b -catenin-mutant cell lines .We evaluated A375,HT-29,HCT 116,colo-205,and SW48cells,which are representative cell lines that are highly sensitive to SMK-17(i.e.,an IC 50of less than 2.0m M),with respect to the effect of SMK-17on the MAPK pathway,cell cycle,and apoptosis induction.SMK-17inhibited ERK1/2phosphorylation in all cell lines in a dose-dependent manner (Figure 2A),as determined by analysis of intracellular MEK1/2activity.Treatment with 1m M SMK-17,which almost completely inhibited ERK1/2phosphorylation,induced G1cell cycle arrest in BRAF-mutant A375and HT-29cells (Figure 2B).However,even at concentrations of up to 10m M,SMK-17did not induce apoptosis in BRAF-mutant A375and HT-29cells,as shown by the increase in the sub-G1population,as determined via flow cytometry (Figure 2B).On the other hand,treatment of cells with SMK-17at the minimum effective concentrations for the inhibition of ERK1/2phosphorylation induced apoptosis in BRAF-mutant colo-205and K-Ras-mutant HCT 116cell lines,as shown by the presence of cleaved PARP (Figure 2A).SMK-17-induced apoptotic cell death wasfurtherFigure 1|Anti-tumor activities of SMK-17in vitro .(A)The chemical structure of SMK-17,its IC 50values for the cell-free kinase reactions of human MEK1and MEK2,and kinase inhibition profiling on 233human kinases at 1000nM of SMK-17.(B)IC 50values for a panel of tumor cell lines harboring several oncogenic mutations treated with the MEK inhibitors,SMK-17and U0126.Cell numbers were determined 72h after compound treatment.IC 50values were calculated from dose-response curves,as described in the Materials and Methods.The figures show the differential growth inhibition pattern of the compounds against the 24tumor cell lines tested.The log y-axis shows the difference between the mean of the log IC 50for the 24cell lines and the log IC 50for each cell line.Values are the mean of three independent experiments.(C)SMK-17and (D)U0126selectively inhibit the proliferation of MAPK-mutated tumor cell lines.Scatter plots show the log IC 50of mutated cell line and the rest one in the MAPK,PI3K,p53,and Wnt pathway (in a left-to-right fashion).P values were obtained by performing a Student’s t -test for group comparisons.confirmed by the detection of a sub-G1population in HCT 116and colo-205cells (Figure 2B).However,the SW48cell line,which does not carry mutations in either the K-Ras or BRAF gene,still underwent apoptosis under conditions in which ERK1/2phosphorylation is inhibited by SMK-17(Figures 2A and 2B).These results indicate that the apoptosis-inducing ability of SMK-17is independent of the status of the MAPK pathway in tumor cells.Therefore,we focused on the differences between the cell lines that underwent either G1arrest or apoptosis by examining the b -catenin status of cells;cell lines that underwent apoptosis after SMK-17treatment commonly harbored a mutation of b -catenin activation.The dependence of SMK-17-induced apoptosis on such mutation was further explored.Other cell lines also harboring such mutation,specifically,SK-MEL-1and LS-174T cells,also underwent apoptosis when treated with SMK-17,as indicated by the increased cell popu-lation at the sub-G1peak (Figure 2C).However,DLD-1and SW620cells,which harbor a K-Ras mutation but not a mutation of b -catenin activation,did not undergo apoptosis under conditions in which the ERK1/2phosphorylation was completely inhibited by SMK-17(Figure 2C).Next,we examined whether another MEK inhibitor alsoselectively induced apoptosis in cell lines harboring activating muta-tions in b -catenin.As shown in Figure 2D,1.0m M PD184352com-pletely inhibited ERK1/2phosphorylation and induced cell cycle arrest at the G1phase in HT29cells.Similarly,PD184352also inhib-ited ERK1/2phosphorylation in DLD-1cells without leading to a significant increase in the sub-G1population (Figure 2D).On the other hand,PD184352inhibited ERK1/2phosphorylation at con-centrations of 0.3m M and higher in HCT 116cells and gradually induced a dose-dependent increase in the sub-G1population.Similar results were obtained in SW48cells,indicating that MEK inhibitors selectively induced apoptosis in cell lines harboring active mutations in b -catenin.Activating mutations in b -catenin are associated with SMK-17-induced apoptosis .To test the hypothesis that active mutations in b -catenin may be responsible for apoptosis mediated by MEK inhibitor,we transfected the active form of b -catenin (S37A,S45A)and EGFP expression plasmids into A375cells harboring wild-type b -catenin and evaluated SMK-17-induced apoptotic cell death by analyzing the levels of cleaved PARP via western blotting.AsFigure 2|MEK inhibitors selectively induced apoptosis in b -catenin-mutated cell lines.(A)SMK-17selectively induced apoptosis in b -catenin-mutated cell lines at the lowest effective concentration.Cells were treated with the indicated concentrations of SMK-17and,after 24h,cell lysates were probed for ERK1/2,phospho-ERK1/2(p-ERK1/2),cleaved PARP,and b -actin expression via western blot.(B)SMK-17induced G1arrest in b -catenin wild-type cells and apoptosis in b -catenin-mutant cells.Cells were treated with the indicated concentrations of SMK-17.After 48h,the sub-G1and G1populations were analyzed by flow cytometry.The percentage of the sub-G1population (red)is indicated in each histogram.(C)SMK-17induced apoptosis in b -catenin-mutant cells.Cells were treated with the indicated concentrations of SMK-17.After 24h,cell lysates were probed for ERK1/2and phospho-ERK1/2(p-ERK1/2)by western blot.After 48h,the sub-G1and G1populations were analyzed by flow cytometry.(D)PD184352induced apoptosis in b -catenin-mutant cells.Cells were treated with the indicated concentrations of PD184352.After 24h,cell lysates were probed for ERK1/2and phospho-ERK1/2(p-ERK1/2)by western blot.After 48h,the sub-G1and G1populations were analyzed by flow cytometry.shown in Figure 3A,expression of the active form of b -catenin did not affect SMK-17-induced inhibition of ERK1/2phosphorylation,although apoptosis was observed after SMK-17treatment in A375cells.To quantitatively detect early apoptosis and late apoptosis induced by SMK-17,active b -catenin/EGFP-transfected A375cells were stained with annexin V/APC and 7-AAD and then analyzed by flow cytometry,with the gates set to include only EGFP-positive cells.As shown in Figure 3B,SMK-17at concentrations of up to 10m M did not affect the proportion of annexin V 1/7-AAD 2cells (early apoptosis)and annexin V 1/7-AAD 1cells (late apoptosis)in control,EGFP-expressing A375cells.On the other hand,the percentage of not only early apoptotic,but also of late apoptotic A375cells expressing the active form of b -catenin increased dose-dependently.At 10m M SMK-17,22.0%of A375cells expressing active b -catenin were early apoptotic,and 46.7%were late apoptotic.In contrast 1.4%of control EGFP-expressing A375cells were early apoptotic and 0.7%were late apoptotic.These results indicate that expression of the active form of b -catenin is required for SMK-17-induced apoptosis.TCF7L2activity is related to apoptosis induced by MEK inhibitor in b -catenin-mutant cell lines .To show that SMK-17-induced apoptosis could be regulated by the enhancement of b -catenin-dependent transcriptional activation,A375cells were co-transfected with the TOPFlash and Renilla luciferase plasmids.Transfected cells were treated with 50ng/mL recombinant Wnt3a for 24h,and their luciferase activity was measured.Wnt3a was found to accelerate TCF7L2transcription in A375cells (Figure 4A).SMK-17-induced apoptosis in untreated and Wnt3a-treated A375cells was analyzed by western blotting for cleaved PARP.We found that levels of cleaved PARP increased after combination treatment with SMK-17and Wnt3a relative to levels after treatment with either agent (Figure 4B).To further confirm that TCF7L2transcriptional activity is involved in SMK-17-induced apoptosis,we next examined the effect of TCF7L2downregulation on SMK-17-induced death of HCT 116cells harboring an active mutation of b -catenin.As shown in Figure 5A,TCF7L2transcriptional activity was successfully reduced after dominant-negative TCF7L2(DN-TCF7L2)plasmid transfection (0.061to 0.033).DN-TCF7L2-transfected cells were then treated with SMK-17and stained with annexin V/APC and 7-AAD for the analysis of apoptosis via flow cytometry.SMK-17-induced apoptosis in control EGFP-expressing HCT 116cells (at 10m M SMK-17,23.2%were annexin V 1/7-AAD 2and 17.1%were annexin V 1/7-AAD 1)was greatly diminished by the expressionofFigure 3|Overexpression of the active form of b -catenin induced apoptosis in SMK-17-treated A375cells.A375cells were transfected with the control vector or with a vector for the active form of b -catenin.Transfected cells were treated with DMSO or SMK-17(1.0,3.0,and 10m M).After 48h,(A)cell lysates were probed for ERK1/2,phospho-ERK1/2(p-ERK1/2),and cleaved PARP by western blot.(B)Cells were stained with annexin V/APC and 7-AAD and then analyzed by flow cytometry,with gates set to include only EGFP-positive cells.Apoptotic cells were defined as being annexinV-positive.Figure 4|Activation of Wnt/b -catenin signaling by Wnt3a induced apoptosis in A375cells.(A)A375cells were co-transfected with the TOPFlash and Renilla luciferase plasmids.Transfected cells were treated with 50ng/mL Wnt3a for 24h,and their luciferase activity was measured.Wnt signaling activity in terms of TCF7L2transcription was monitored by using the TOPFlash/continuous Renilla luciferase assay.The ratio of the firefly luciferase intensity of TOPFlash to that of Renilla luciferase was used as an indicator of TCF7L2transcriptional activity.(B)Cells were treated with either DMSO or with 10m M SMK-17,with or without 50ng/mL recombinant Wnt3a,for 48h.Cell lysates were probed for ERK1/2,phospho-ERK1/2(p-ERK1/2),and cleaved PARP via western blot.Cleaved PARP was observed after combination treatment with SMK-17and Wnt3a.DN-TCF7L2(at 10m M SMK-17,14.8%were annexin V 1/7-AAD 1and 3.1%were annexin V 1/7-AAD 1;Figure 5B).Thus,SMK-17-induced apoptosis positively correlated with TCF7L2transcriptional activity.Involvement of c-Myc-mediated apoptosis gene expression in SMK-17-induced apoptosis of b -catenin-mutant cells .As b -catenin-mediated increase in TCF7L2transcriptional activity is involved in SMK-17-induced apoptosis,we next examined the possibility that SMK-17-induced apoptosis in b -catenin-mutant cells is mediated by c-Myc,whose promoters are directly activated by TCF7L2.To do this,HCT 116cells were transiently transfected with c-myc siRNA,and their cell viability following SMK-17treatment was examined.As shown in Figure 6A,SMK-17dose-dependently decreased both c-Myc and phosphorylated ERK1/2levels (Figure 6A).Furthermore,as the PARP cleavage and sub G1population detected by flow cytometry analysis show,silencing c-Myc expression suppressed SMK-17-induced apoptosis.Indeed,as shown in Figure 6B and 6C,Wnt3a-induced PARP cleavage and sub G1population in SMK-17-treated A375cells were also inhibited by the knockdown of c-Myc.These results indicate that c-Myc is a key regulator involved in apoptosis induced by MEK inhibitor when the Wnt pathway is activated,for example,during Wnt3a stimulation or when b -catenin is mutated.Apoptosis induction of SMK-17in b -catenin-mutated xenograft models .Because SMK-17selectively induced apoptosis in cell lines harboring activating mutations in b -catenin in vitro ,we next examined the ability of SMK-17to induce apoptosis in vivo .Active b -catenin-mutant tumor cells (SW48and colo-205cells)and wild-type b -catenin tumor cells (A375and HT-29cells)were injected subcutaneously into NOD-SCID mice (SW48cells)or nude mice (other cell lines)to establish xenograft models.SMK-17or the control vehicle was then administered by oral gavage.We confirmed the inhibitory activity of SMK-17toward intratumor MEK by monitoring levels of phosphorylated ERK1/2in SW48and A375cells in vivo after a single dose of SMK-17(Supplemental Figure S1).As shown in Figure 7A,tumor regression in response to multiple daily oral administration of SMK-17at half MTD (200mg/kg)was observed only in active b -catenin-mutant xenograft models,namely,models using SW48and colo-205cells.On the other hand,the maximum effect of SMK-17at MTD dosing (400mg/kg)was just growth inhibition of A375and HT-29cells expressing wild-type b -catenin,without significant body weight loss.TUNEL staining revealed significant apoptosis induction in SW48tumor tissues from SMK-17-treated but not in SW48tumor tissues from control mice (Figure 7B).In contrast,apoptotic cells were not observed in the SMK-17-treated A375xenograft model.DiscussionSMK-17is a highly selective inhibitor of MEK1and MEK2in a non-ATP-competitive manner,possibly because of its binding to the allosteric pocket of MEK1/217.In addition,we reported that SMK-17inhibited cell growth in colon-26and HT-29cell lines harboring highly phosphorylated MEK1/2and ERK1/217.In this study,we investigated the effect of SMK-17on several types of human tumor cells in detail.We found SMK-17to be highly effective in limiting the proliferation of tumor cells with aberrant activated MAPK pathway signaling (Figures 1B and 1C).Furthermore,tumor cells harboring BRAF mutations were found to be more sensitive to SMK-17than were tumor cells with K-Ras mutations.This is similar to results for another MEK inhibitor 18.These observations can be explained by the fact that BRAF mutations could directly influence MEK activity,as it is an immediate upstream effector of MEK,whereas Ras mutations activate additional signaling pathways that bypass MEK 19.Thus,BRAF mutations predict a cell’s sensitivity to MEK inhibition 19.Interestingly,SMK-17-sensitive BRAF-and K-Ras-mutant cells could be classified into two groups based on the fate of cells after SMK-17treatment.One contains tumor cells that undergo SMK-17-induced cell-cycle arrest (BRAF-mutant A375and HT-29cells and K-Ras-mutant SW620cells),and the other consists of tumor cells that undergo apoptosis when SMK-17inhibits the phosphorylation of ERK1/2(BRAF-mutant colo-205cells and K-Ras-mutant HCT 116and LS-174T cells).Therefore,it is difficult to predict whether SMK-17elicits cell growth inhibition or apoptosis solely on the basis of their BRAF or K-Ras mutation status.However,we also found that their sensitivity to the MEK inhibitor was closely related to the status of mutations in canonical Wnt/b -catenin signaling (Figure 1C).Indeed,we found that a common feature of tumor cells that undergo apoptosis following SMK-17treatment is the presence of an active mutation in b -catenin.Mutations in b -catenin in these cells involve the deletion or the exchange of serine and threonine residues at positions 45and 33(HCT 116cells:S45deletion,SW48cells:S33Y,LS-174T cells:S45F,and SK-MEL-1cells:S33C),thus interfering with itsefficientFigure 5|Expression of DN-TCF7L2reduced SMK-17-induced apoptosis in HCT 116cells.HCT 116cells were transfected with DN-TCF7L2.(A)Luciferase activity was monitored through co-transfection with the TOPFlash and Renilla luciferase plasmids.(B)HCT 116cells expressing DN-TCF7L2were treated with the indicated concentrations of SMK-17for 48h and stained with annexin V/APC and 7-AAD.Cells were analyzed by flow cytometry with gates including only EGFP-positive cells.ubiquitination and degradation by the proteasome 20.Furthermore,colo-201and colo-205cells showed a homozygous A-to-G missense transition mutation at codon 287in exon 6,resulting in the substi-tution of serine for asparagine (N287S).Therefore,among the tumor cells tested,only active b -catenin mutants,irrespective of the status of BRAF or K-Ras mutation,underwent apoptosis after treatment with the MEK inhibitor at minimum effective concentrations for the inhibition of ERK1/2phosphorylation.A requirement for an active b -catenin mutation for SMK-17-induced apoptosis was confirmed by the following findings:1)SMK-17induced apoptosis in A375cells expressing the active form of b -catenin and harboring wild-type b -catenin (Figure 3A and 3B).2)Stimulation of Wnt/b -catenin signaling with Wnt3a enhanced the ability of SMK-17to induce apoptosis in A375cells (Figures 4A and 4B).3)Inhibition of b -catenin signaling by the expression of DN-TCF7L2suppressed SMK-17-induced apoptosis in HCT 116cells harboring an active b -catenin mutation (Figures 5A and 5B).These results are consistent with the findings of another study show-ing that apoptosis mediated by targeted BRAF inhibition in mela-noma is dependent on b -catenin 21.What remains unclear is the mechanism by which SMK-17selec-tively induced apoptosis in cell lines harboring active,mutated b -catenin.Through interaction with TCF/LEF transcription factors,b -catenin promoted the expression of Wnt target genes such as those for c-Myc ,cyclin D2,and CD4422.There is now strong evidence that increased c-Myc expression is a key component of Wnt signaling,which regulates the expression of genes involved in diverse cellularprocesses such as apoptosis,cell cycle progression,cell growth,and DNA replication 23,24.Furthermore,c-Myc protein is known to be phosphorylated and stabilized by ERK1/225;indeed,inhibition of ERK phosphorylation by SMK-17led to decreased c-Myc expression (Figure 6A).Nevertheless,we confirmed that c-Myc leads to SMK-17-induced apoptosis in tumor cells harboring b -catenin mutations;c-Myc knockdown inhibited SMK-17-induced apoptosis not only in b -catenin-mutant HCT 116cells,but also in Wnt3a-stimulated A375cells.We also determined which c-Myc-mediated,apoptosis-related proteins contributed to SMK-17-induced apoptosis in tumor cells harboring b -catenin mutations.Several studies demonstrated the involvement of BIM in apoptosis induced by RAF or MEK inhib-itor 21,26–31.BIM,the Bcl-2homology domain 3-only (BH3-only)pro-tein,is known to bind and inhibit prosurvival Bcl-2family members.Expression of BIM is regulated by c-Myc 26,32and is inhibited by BRAF-MEK-ERK signaling in mouse and human melanocytes,as well as in human melanoma cells 33.Therefore,inhibition of BRAF-MEK-ERK signaling increases the levels of BIM expression,inducing cells to undergo apoptosis.We also found that SMK-17induced an increase in BIM expression levels upon inhibition of ERK phosphor-ylation in b -catenin-mutant HCT 116,SW48,and colo-205cells (Supplemental Figure S2).However,knockdown of BIM failed to suppress SMK-17-induced apoptosis in all cell lines tested (data not shown).Therefore,proteins related to c-Myc-mediated apopto-sis other than BIM might be responsible for the active b -catenin-mediated apoptosis induced by SMK-17.Furthermore,consistentFigure 6|Involvement of c-Myc in SMK-17-induced apoptosis.(A)HCT 116cells were transfected with control or c-myc siRNA.Transfected cells were treated with the indicated concentrations of SMK-17for 24h.Western blotting of cell lysates showed reduced expression of phospho-ERK1/2(p-ERK1/2)and c-Myc protein and increased expression of cleaved PARP.(B)A375cells were transfected with control or c-myc siRNA.Transfected cells were treated with the indicated concentrations of SMK-17in the presence or absence of 20ng/mL Wnt3a for 24h.The expression of ERK1/2,p-ERK1/2,c-Myc,active b -catenin,and cleaved PARP was detected by western blotting.(C)A375cells were transfected with control or c-myc siRNA.Transfected cells were treated with the indicated concentrations of SMK-17in the presence or absence of 20ng/mL Wnt3a for 48h.The apoptosis induction was detected by flow cytometry analysis after cytological staining using PI.with the previous report 34,c-Myc protein expression in b -catenin mutated tumor cells seems to be higher than in b -catenin/APC wild-type tumor cells (Supplemental Figures S3A &B).On the other hand,the amount of c-Myc mRNA seemed to not correlate with the tran-scriptional activity of TCF7L2(Supplemental Figure S3C).Moreover,c-Myc protein expression had no increase in level in Wnt3a-stimulated A375cells (Figure 6B).Therefore,the link between TCF7L2and c-Myc is still controversial,and although knockdown of c-Myc inhibited SMK-17-induced apoptosis,we can-not exclude the possibility that another target protein of the canon-ical Wnt pathway apart from c-Myc is responsible for apoptosis induced by the MEK inhibitor.In this study,we used three APC-mutant colorectal tumor cells,namely,SW480,SW620,and DLD-1cells.SW480and SW620cells have a mutant version of the APC tumor suppressor gene,which causes premature termination of the protein at amino acid 13335,36.Codon 1416of the APC gene is mutated,and the other allele is lost in DLD-1cells,resulting in constitutively active TCF/LEF 37.b -catenin and APC mutations appear to be mutually exclusive,possibly reflect-ing the fact that both components act in the same pathway.Nevertheless,SMK-17induced cell death in SW480cells and in b -catenin-mutant cells,but not in SW620or DLD-1cells (Supplemental Figure S4).SW480and SW620cells are two colon tumor cell lines established from the same patient although they have different metastatic potential 38.They are well-characterized and rep-resent the differing features of primary and metastatic sites 39.Indeed,we found that expression levels of survivin,an anti-apoptotic protein,was significantly higher in SW620than in SW480cells (Supplemental Figure S4);this difference in survivin expression levels may explain why SW620but not SW480cells are resistant to SMK-17-induced apoptosis.On the other hand,SMK-17did not inhibit cell growth or apoptosis in DLD-1cells.DLD-1cells expressed high amounts of ERK1/2protein,although its phosphor-ylation level was quite low (Supplemental Figure S4).Therefore,it is possible that proliferation of DLD-1cells is independent of the MEK/MAPK pathway.Thus,although we do not know at present why b -catenin mutations,but not APC mutations,could predict a cell line’s sensitivity to MEK inhibitors,we found that TCF7L2transcriptional activity was higher in APC mutants than in b -catenin mutants (Supplemental Figure S5).This difference may affect the balance of c-Myc-induced expression of pro-apoptotic and anti-apoptotic pro-tein,thus regulating tumor cell survival.Therefore,moderatelyenhanced TCF7L2activity that is regulated by b -catenin mutations might be required for apoptosis induced by MEK inhibitor.Another possible explanation is that although tumor initiation by either loss of APC function or oncogenic b -catenin mutations is functionally equi-valent,the APC gene may have other functions,namely the ability to capture the kinetochore in the establishment of the mitotic spindle,a loss which underlies malignant progression,therefore,mutations in APC are responsible for chromosomal instability,and are independ-ent of their roles in Wnt signal transduction 40.Chromosome instab-ility will then confer a mutant phenotype,allowing further malignant progression through numerical and structural chromosomal rear-rangements.Thus APC mutation will accumulate mutations in other genes including pro-and anti-apoptotic genes,allowing cells to be resistant to MEK inhibitors.Thus,because of the diverse phenotypes produced by gene instability,it appears challenging to predict the sensitivity of APC mutant cells to MEK inhibitors.To link these findings to a clinically relevant model,we conducted in vivo studies using active b -catenin-mutant xenograft models.Significant tumor regression without any severe toxicity was observed in b -catenin-mutant SW48and colo-205cells in response to daily oral administration of SMK-17(Figures 7A and 7B).At present,MEK inhibitors have been evaluated in both preclinical and clinical studies as a potential therapeutic option for patients with melanomas carrying the BRAF V600-mutation;however,single-agent efficacy has been limited.Therefore,rational combination strategies using MEK inhibitors with other molecularly targeted drugs are expected to lead to greater efficacy.For example,several studies have reported crosstalk between Wnt/b -catenin and MAPK signaling 41,42.Accordingly,the combination of MEK blockade and Wnt pathway modulation has shown synergistic anti-proliferative effects in both in vitro and in vivo preclinical colorectal tumor models 43.However,we report in this study that a MEK inhibitor alone could induce signifi-cant tumor regression in b -catenin-mutant xenograft models.As b -catenin is mutated in up to 10%of all sporadic colon carcinomas resulting from point mutations or in-frame deletions of serine and threonine residues phosphorylated by GSK3b 44,our findings support the clinical use of MEK inhibitors as single agents for patients with colorectal carcinoma carrying active b -catenin mutations.On the other hand,studies with patient cohorts have been reported to determine whether active Wnt signals correlate with clinical responses to the BRAF inhibitor,and it was reported that increased nuclear b -catenin in biopsies is associated withdecreasedFigure 7|Antitumor activities of SMK-17in vivo .(A)SMK-17induced tumor regression and apoptosis in b -catenin-mutated cell lines in vivo .SMK-17suspensions and the vehicle control (0.5%MC)were orally (po)administered at 10mL/kg once daily.(B)Apoptosis induced by SMK-17in tumors.TUNEL staining results were obtained from tumors from xenograft mice 24h after the final oral administration of SMK-17(q.d.32days).Each indicated score (%)was the mean ratio of the apoptotic cells in the tissue slices from 4independent tumor-bearing mice.。
【国家自然科学基金】_gsk3β_基金支持热词逐年推荐_【万方软件创新助手】_20140729
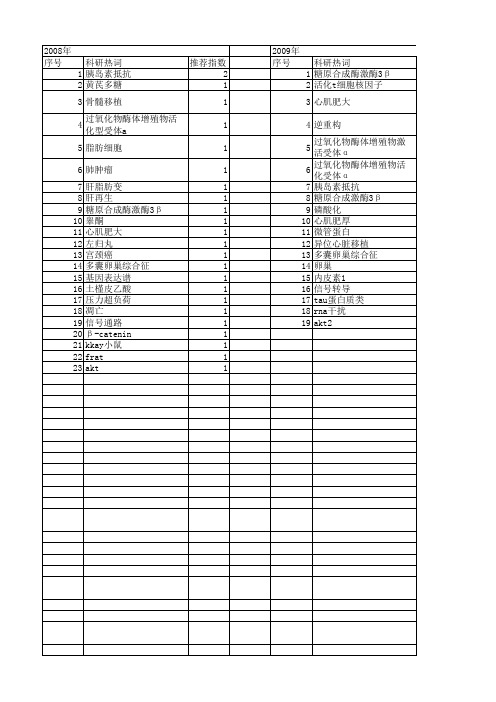
推荐指数 3 3 2 2 2 2 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
小分子药物 1 学习记忆 1 基因克隆 1 哌立福辛 1 吲哚美辛 1 分子作用机制 1 七氟醚 1 β -catenin 1 α -synuclein 1 wnt信号通路 1 tcf3 1 tau蛋白 1 rna干扰 1 pyrvinium 1 pc12细胞 1 notch信号 1 n-乙酰基-丝氨酰-天门冬酰-赖氨酰-脯氨酸 1 mgc-803 1 mek 1 lif 1 hbx蛋白 1 gst-gsk3β 1k85m 1 gst-gsk3β 1 gsk3β 基因 1 gsk3β /β -catenin 1 gsk3β 1 gsi 1 c-myc 1 akt/gsk3β /nag-1通路 1
2008年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 2 黄芪多糖 1 骨髓移植 1 过氧化物酶体增殖物活化型受体a 1 脂肪细胞 1 肺肿瘤 1 肝脂肪变 1 肝再生 1 糖原合成酶激酶3β 1 睾酮 1 心肌肥大 1 左归丸 1 宫颈癌 1 多囊卵巢综合征 1 基因表达谱 1 土槿皮乙酸 1 压力超负荷 1 凋亡 1 信号通路 1 β -catenin 1 kkay小鼠 1 frat 1 akt 1
2013年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52
Wnt 、β-Catenin通路促进替代活化型巨噬细胞的激活将加重肾纤维化

在BMM中,IL-4或TGFβ1刺激的巨噬细胞M2极化被ICG-001处理显着抑制。 我们还从小鼠分离的BMM以诱导β-catenin蛋白基因缺失, β-catenin在BMM 中的消除可以阻止IL-4或TGFβ1刺激的巨噬细胞M2极化。
为了探讨β-catenin调节巨噬细胞M2极化的机制,我们检测了两种细 胞类型中STAT3在Y705的磷酸化。
为了探讨STAT3激活在Wnt3a加重巨噬细胞M2极化中的作用,我们用STAT3抑制剂 Stattic处理BMMs。结果表明,Stattic可以很大程度上抑制STAT3磷酸化和IL-4或 TGFβ1加Wnt3a诱导的巨噬细胞M2极化,表明STAT3激活促进巨噬细胞M2极化的 关键作用。
因此,可以得出结论,Wnt3a通过上调STAT3表达和活化来加剧IL-4-或TGFβ1诱导 的巨噬细胞M2极化。
Wnt信号传导被分为经典和非经典信号通路,取决于β-catenin是否被激 活。对于经典的Wnt信号通路,β-catenin在激活后积累在细胞质中,然 后转移到细胞核中,结合细胞因子并刺激参与组织纤维形成的靶基因的 转录。
Wnt / β-catenin激活可以防止小鼠肾小管细胞死亡和AKI。然而,足细胞 或肾小管细胞中Wnt / β-catenin的异常激活可能加重蛋白尿,肾功能不 全和肾纤维化。此外,β-catenin可以调节髓系细胞的运动性和粘附性, 这有助于皮肤损伤后间充质细胞的积累。因此,有人推测在肾纤维化中, Wnt / β-catenin活化可能促进巨噬细胞迁移并促进其累积。最近据报道, β-catenin相关信号在肺部髓系细胞中的激活导致肺泡巨噬细胞亚型的分 化,其拮抗肺纤维化的消退。强制表达β-catenin变体能够促进巨噬细胞 分化。在这方面,Wnt / β-catenin信号可能调节巨噬细胞的积累和活化 并加重CKD中的肾纤维化。
GSK-3β、Snail和E-cadherin在三阴乳腺癌组织中的表达及临床意义

GSK-3β、Snail和E-cadherin在三阴乳腺癌组织中的表达及临床意义高双全;肖高芳;高双荣;王林辉;丁宇;杜日昌【摘要】目的探讨GSK-3β、Snail和E-cadherin在三阴乳腺癌(TNBC)中表达及意义.方法选择2006年9月至2013年9月收治的48例TNBC患者作为观察对象即TNBC组,并随机选出60例非TNBC患者作为对照即NTNBC组,应用免疫组织化学SP法检测GSK-3β、Snail和E-cadherin的表达情况,并进行统计分析.结果 TNBC组GSK-3β、Snail的阳性表达率显著高于NTNBC组(P<0.05),E-cadherin的阳性表达率显著低于NTNBC组(P<0.05).GSK-3β与Snail表达呈正相关(P<0.05),与E-cadherin表达呈负相关(P<0.05).结论Snail、GSK-3β、E-cadherin可能共同参与乳腺癌上皮间质转化过程,可成为综合评价TNBC的重要标志物.【期刊名称】《重庆医学》【年(卷),期】2015(044)021【总页数】3页(P2884-2886)【关键词】三阴乳腺肿瘤;糖原合成激酶-3β;蜗牛蛋白;E-钙粘蛋白【作者】高双全;肖高芳;高双荣;王林辉;丁宇;杜日昌【作者单位】韶关市粤北人民医院病理科,广东韶关512026;韶关市粤北人民医院病理科,广东韶关512026;中国中医科学院中药研究所,北京100029;韶关市粤北人民医院病理科,广东韶关512026;韶关市粤北人民医院病理科,广东韶关512026;韶关市粤北人民医院病理科,广东韶关512026【正文语种】中文【中图分类】R365三阴乳腺癌(triple negative breast cancer,TNBC)表现为雌激素受体(estrogen receptor,ER)、孕激素受体(progesterone receptor,PR)和人表皮生长因子受体2(human epidermal growth factor receptor-2,Her-2)表达均缺失的一种特殊类型的乳腺癌[1],占全部乳腺癌的13.7%~23.8%[2],具有发病年龄轻、侵袭性高、易复发转移、生存期短及预后差等特点,严重威胁了患者的生命。
磷脂酰肌醇3激酶β(PI3Kβ)和PI3Kδ在KIT突变介导的细胞转化中起不同作用
细胞与分子免疫学杂志(Chin J Cell Mollmmunol)2021,37( 1)39•论著•文章编号:1007-8738(2021 )01>0039~08磷脂酰肌醇3激酶p(PI3Kp)和PI3K8在K IT突变介导的细胞转化中起不同作用张少婷,朱光荣,石君,杨继辉,蒋宗英,张良颖,窦凯凯,孙建民*(宁夏医科大学基础医学院病原生物学与医学免疫学系,宁夏银川750004)[摘要]目的探究磷脂酰肌醇3激酶(P I3K)的不同亚型在三型酪氨酸激酶受体KIT突变介导的信号传递及细胞增殖中的作 用。
方法在B aF3细胞中稳定表达野生型KIT及胃肠间质瘤中常见的KIT突变V560D、W557K558del,分别用PI3K a、P I3KP、P I3K5亚型特异性抑制剂或者广谱P I3K抑制剂处理细胞,免疫沉淀法和W estern blot法检测KIT及其下游信号活化情况。
胃肠 间质瘤G IST-T1细胞采用相同药物及浓度处理,免疫共沉淀和W estern blot法检测KIT及其下游信号的活化情况,噻唑蓝 (M T T)法检测细胞增殖,流式细胞术检测细胞凋亡。
结果与对照组相比,在表达野生型KIT及其突变体的B aF3细胞中,P I3K8亚型特异性抑制剂对KIT及其下游信号分子蛋白激酶B(AKT)和胞外信号调节激酶(ERK)活化的抑制作用最强,其次为 P I3K a和P I3KP亚型特异性抑制剂。
在G IST-T1细胞中,P I3KP亚型特异性抑制剂对KJT及其下游信号活化的抑制作用最强,其次为P I3K&和P I3K a亚型特异性抑制剂。
结论在B aF3细胞中,P I3KS亚型在KIT活化及其下游信号传递中起主要作用,而在G IST-T1细胞中,P I3Kp亚型在KIT活化及其下游信号传递中起主要作用,这些结果表明不同P I3K亚型在KIT突变介导 的细胞转化中起不同作用,且在不同的细胞中其作用也有不同。
灵芝三萜通过Wnt信号通路对胶质瘤细胞侵袭和迁移的影响
灵芝三萜通过Wnt信号通路对胶质瘤细胞侵袭和迁移的影响
王炳清,马庆波,刘敬博,高俊波(北大医疗鲁中医院神经外科,山东淄博 255400)
[摘 要]目的 分析灵芝三萜通过Wnt信号通路对胶质瘤细胞侵袭和迁移的影响。方法 用10mg/L(低剂
量组)、50mg/L(中剂量组)、100mg/L(高剂量组)的灵芝三萜处理人胶质瘤U251细胞,并设置对照组(未给予灵芝三萜处理);比较处理不同时刻及不同浓度灵芝三萜对胶质瘤细胞U251的增殖、侵袭、迁移及凋亡的影响,同时检测U251细胞中β连环蛋白(βcatenin)、磷酸化糖原合成酶激酶3β(pGSK3β)蛋白的表达水平。结果 药物处理不同时刻对照组U251细胞的OD值、侵袭细胞数、迁移率、凋亡率、βcatenin、pGSK3β蛋白表达均高于3个灵芝三萜组,差异有统计学意义(P<005);随着处理灵芝三萜浓度的升高,U251细胞的OD值、侵袭细胞数、迁移率、凋亡率均逐渐降低,βcatenin、pGSK3β蛋白表达均逐渐升高,差异有统计学意义(P<005);随着处理时间的延长转染同浓度的灵芝三萜U251细胞的OD值、侵袭细胞数、迁移率、凋亡率均升高,βcatenin、pGSK3β蛋白表达均降低,差异有统计学意义(P<005)。结论 灵芝三萜可能够通过Wnt信号通路抑制胶质瘤细胞的增殖、侵袭和迁移,促进细胞凋亡。[关键词]胶质瘤;灵芝三萜;Wnt/β连环蛋白信号通路;增殖;侵袭;迁移;凋亡
[中图分类号]R2855 [文献标识码]A [文章编号]20968388(2021)09103106
DOI:10.19367/j.cnki.20968388.2021.09.007
EffectsoftriterpenesofganodermalucidumontheinvasionandmigrationofgliomacellsthroughWntsignalingpathway
β-catenin在胃癌中的表达及其机制
β-catenin在胃癌中的表达及其机制王海波;刘延军;陈树伟;武文龙;李凤臣【摘要】目的探讨Wnt信号通路中心调控分子β-catenin在胃癌中的表达及其在胃癌发病中的作用机制.方法利用shRNA病毒沉默BGC-823胃癌细胞株中的β-catenin的表达,利用空载病毒感染细胞株作为对照组,实时荧光定量PCR及Western blot检测病毒沉默效果,将转染后的细胞株注入裸鼠颈部构建裸鼠胃癌模型,每周检测2组裸鼠皮下肿瘤生长情况并测量记录皮下瘤块直径.成瘤裸鼠模型饲养8w后分离完整皮下瘤块,统计瘤块数量及瘤块重量.Western blot检测2组裸鼠皮下瘤块中Wnt信号通路相关分子DKK1、c-myc、CyclinD1的表达变化.结果β-catenin沉默后的BGC-823胃癌细胞株注射裸鼠,其皮下瘤块的数量、直径以及重量均较对照空载病毒转染的BGC-823胃癌细胞株注射的裸鼠少,差异具有统计学意义(P<0.05),且β-catenin沉默组裸鼠,其瘤块中DKK1、c-myc、CyclinD1的表达水平较对照组裸鼠显著降低(P<0.05).结论β-catenin通过上调DKK1、c-myc、CyclinD1的表达促进胃癌细胞的增殖,因此β-catenin在胃癌发生发展过程中具有重要作用,其可能作为评估胃癌预后的诊断指标.【期刊名称】《中国生化药物杂志》【年(卷),期】2016(000)007【总页数】4页(P32-35)【关键词】β-catenin;胃癌;Wnt信号通路;BGC-823胃癌细胞株【作者】王海波;刘延军;陈树伟;武文龙;李凤臣【作者单位】解放军第107医院胃肠外科,山东烟台264002;解放军第107医院胃肠外科,山东烟台264002;解放军第107医院胃肠外科,山东烟台264002;解放军第107医院胃肠外科,山东烟台264002;解放军第107医院胃肠外科,山东烟台264002【正文语种】中文【中图分类】R735.2胃癌是全球高发的恶性肿瘤之一,尤其是在广大的农村地区,由于饮食卫生等因素的影响,胃癌全球每年新发病例已增至150万[1- 2]。
Gsk-3ββ-catenin CyclinD1在脑胶质瘤中的表达及意义
Gsk-3ββ-catenin CyclinD1在脑胶质瘤中的表达及意义张勇;王斌;周幽心【期刊名称】《基层医学论坛》【年(卷),期】2014(000)001【摘要】目的:探讨Gsk-3β、β-catenin和CyclinD1在脑胶质瘤中的表达及意义。
方法应用免疫组化Envision法检测Gsk-3β、β-catenin和CyclinD1在48例胶质瘤和11例对照组(正常脑组织)中的表达情况。
结果Gsk-3β、β-catenin、CyclinD1的表达在脑胶质瘤和对照组(正常脑组织)之间以及胶质瘤的高低级别组之间差异均具有统计学意义(P<0.05),β-catenin和CyclinD1在颅内胶质瘤中的表达呈正相关(P<0.05),β-catenin与Gsk-3β在胶质瘤中的表达呈负相关(P<0.05)。
结论胶质瘤组织中存在Gsk-3β低表达和β-catenin高表达,以及细胞周期调节紊乱,这三种蛋白可能在胶质瘤的发生过程中起一定作用。
【总页数】3页(P14-16)【作者】张勇;王斌;周幽心【作者单位】淮北市人民医院,安徽淮北 235000;淮北市人民医院,安徽淮北235000;苏州大学附属第一医院江苏苏州 215006【正文语种】中文【相关文献】1.GSK-3β和β-catenin在弥漫性大B细胞性淋巴瘤组织中的表达及意义 [J], 叶春美;孙爱宁;2.GSK-3β和β-catenin在弥漫性大B细胞性淋巴瘤组织中的表达及意义 [J], 叶春美;孙爱宁3.β-catenin、GSK-3β在不同分子分型乳腺癌中的表达及意义 [J], 宫国良;陈静宁;刘晓雨;刘志军4.β-catenin、CyclinD1与Ki-67在人脑胶质瘤中的表达及意义 [J], 刘婷;孙振柱5.β-catenin、 CyclinD1、c-myc蛋白在脑胶质瘤中的表达及意义 [J], 潘旭波;徐振光;姜蕾;王威;于文芳因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Toxicology Letters 222 (2013) 212–223Contents lists available at ScienceDirectToxicologyLettersj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /t o x l etGSK3/-catenin signaling is correlated with the differentiation of glioma cells induced by wogonin ଝYajing Wang a ,1,Yi Zhang a ,1,Chen Qian a ,Min Cai a ,Yan Li b ,Zhiyu Li c ,Qidong You c ,Qing Wang d ,Rong Hu a ,∗,Qinglong Guo a ,∗aState Key Laboratory of Natural Medicines,Jiangsu Key Laboratory of Carcinogenesis and Intervention,Department of Physiology,China Pharmaceutical University,24Tongjiaxiang,Nanjing 210009,PR China bJiangsu Centers for Diseases Prevention and Control,Nanjing 210009,PR China cDepartment of Medicinal Chemistry,China Pharmaceutical University,Nanjing 210009,PR China dDepartment of Neurosurgery,Wuxi Second Hospital Affiliated Nanjing Medical University,Wuxi 214002,PR Chinah i g h l i g h t s•Wogonin inhibited C6cells growthand arrested at G0/G1phase of the cell cycle.•Wogonin induced typical morpho-logical changes and an increase in GFAP level.•Induction of cell differentiation by wogonin ascribed to the restrain of GSK-3/-catenin pathway.•Blocking GSK-3/-catenin signaling promotes the glial differentiation of glioma cells.g r a p h i c a la b s t r a cta r t i c l ei n f oArticle history:Received 3February 2013Received in revised form 9July 2013Accepted 10July 2013Available online 19 July 2013Keywords:WogoninGlioblastoma cells DifferentiationGSK-3/-catenin pathway Cell cyclea b s t r a c tMalignant gliomas are the most common and most aggressive primary brain tumor,and for which dif-ferentiation therapy has emerged as a promising candidate strategy.In this study,we used in vitro and in vivo assays to examine the differentiation effects of wogonin,a major active constituent of Scutellaria baicalensis ,on glioma C6and U251cells.We found that wogonin can suppress cell proliferation and induce G0/G1arrest under a concentration-dependent manner.Wogonin also triggered significant reduction in the G1cell-cycle regulatory proteins cyclin D1,cyclin-dependent kinase 2and 4along with overexpress-ion of cell-cycle inhibitory proteins p27.Immunofluorescence and western blot analysis indicated that wogonin increased the expression of lineage-specific differentiation marker glial fibrillary acidic protein (GFAP).In mechanisms,we verified that wogonin significantly diminished the phosphorylated level of protein kinase B (AKT),and maintenance of low -catenin expression level was dependent on glyco-gen synthase kinase 3(GSK3)activation at Ser9.Blocking GSK3/-catenin pathway was required for wogonin-induced proliferation inhibition and terminal differentiation by using canonical activator lithium chloride (LiCl)and inhibitor dickkopf-1(Dkk1).Moreover,intravenous administration of wogonin delayed the growth of C6glioma in the intracranial tumor model.These findings provide the evidence and mechanistic support for wogonin-based differentiation therapies for malignant glioblastoma.Further-more,inhibition of GSK3/-catenin pathway may be a key and requisite factor in glioma differentiation.© 2013 The Authors. Published by Elsevier Ireland Ltd. All rights reserved.ଝThis is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License,which permits non-commercial use,distribution,and reproduction in any medium,provided the original author and source are credited.∗Corresponding authors.Tel.:+862583271055;fax:+862583271055.E-mail addresses:michelhu@ (R.Hu),anticancer drug@ (Q.Guo).1These two authors contributed equally to the manuscript.0378-4274/$–see front matter © 2013 The Authors. Published by Elsevier Ireland Ltd. All rights reserved./10.1016/j.toxlet.2013.07.013Y.Wang et al./Toxicology Letters222 (2013) 212–2232131.IntroductionGlioblastoma multiforme(GBM)is the most common primary tumor of the brain and one of the most lethal malignant tumors, whose highly invasion and metastasis leave rare opportunity for cure.In spite of advances in surgery,optimized radiotherapy and chemotherapy,patients face a dismal prognosis and the median survival rates range from12to15months(Louis et al.,2007). Therefore,great efforts have been made to develop novel adjuvant therapeutic strategies.Differentiation therapy,an approach to the treatment of advanced or aggressive malignancies,involves using agents,alone or in combination,that promote cancer cells maturation and dif-ferentiation into mature cells(Leszczyniecka et al.,2001).This therapeutic protocol is based on the concept that cancer cells dis-play alterations in differentiation states and growth control that can be corrected by appropriate differentiation inducers.More and more studies confirm that this therapy is effective in the treatment of several types of cancer cells.Thefirst differenti-ation agent found to be successful was all-trans-retinoic acid (ATRA)in the treatment of acute promyelocytic leukemia(APL), demonstrating the remarkable efficient to induce glioma cells dif-ferentiation(Bianchi et al.,2008;Kiningham et al.,2008;Redova et al.,2010;Siddikuzzaman Guruvayoorappan and Berlin Grace, 2011;Zhang et al.,2008d).Subsequently,several differentiation-inducing agents,including cholera toxin,capsaicin,valproic acid, neomycin,HMBA,saikosaponins,were reported to be capable of inhibiting cells growth and inducing glioma cells differentiation (Benitez et al.,2008;Cuevas et al.,2004;Gil and Kang,2008; Shirsat et al.,2001;Tsai et al.,2002;Xu et al.,2009).In addition, neuroblastoma cells exhibited effective differentiation with eleva-tion of intracellular adenosine3 ,5 -cyclic monophosphate(cAMP) concentration after treatment with cyclic nucleotide phosphodi-esterase inhibitors or adenylate cyclase activators(Chang et al., 2011;Hai et al.,2009).Although the roles of differentiation inducers obtained growing concern,effective differentiation agents for the treatment of malignant glioma have not yet been found(Kawamata et al.,2006).Wogonin,5,7-dihydroxy-8-methoxyflavone,is a naturally monoflavonoid extracted from Scutellaria baicalensi Georgi,which has been shown to have anti-inflammatory,anticancer,antivi-ral and neuroprotective effects.We have previously reported that wogonin could suppress promyelocytic leukemia U-937cells growth,inducing the granulocytic differentiation and G1arrest of U-937and NB4cells by up-regulating the expression of PLSCR1 gene(Zhang et al.,2008a,b).However,the potential effect of wogo-nin on the induction of solid tumors differentiation has not yet been explored.In addition,a recent study has indicated that wogonin can induce differentiation of neural precursor cells into neuronal cells both in culture and in vivo(Lim et al.,2010).These data prompt us to investigate the possible roles of wogonin on glioma cells differ-entiation.In this study,we investigated the effects of wogonin on cell survival and differentiation in glioma C6and U251cells and explored the underlying molecular mechanisms,in order tofind the potential therapeutic function of wogonin in the treatment of ing a syngeneic rat glioma tumor model,com-prising orthotopic transplantation of C6glioma cells into SD rats,we reported delayed growth of tumor following intravenous administration of wogonin.And we also examined whether the glioma C6cells pretreated with wogonin prior to transplanta-tion into the adult rat brain can be efficiently differentiated into mature astrocytes cells.Our results demonstrated that wogonin effectively induced G0/G1phase cell cycle arrest and the dif-ferentiation of glioma cells focusing on the GSK-3/-catenin signaling.2.Materials and methods2.1.Chemicals and antibodiesWogonin(purity>99%)was isolated from S.baicalensis Georgi according to the procedure reported previously(Hui et al.,2002).Stock solutions of wogo-nin were dissolved in dimethyl sulfoxide(DMSO),stored at20◦C and diluted with medium until needed.Thefinal DMSO concentration in all drug-treated cells was<0.1%.All-trans retinoic acid(ATRA,used as positive control),DAPI, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT)were all pur-chased from Sigma–Aldrich(St.Louis,MO,USA).Stock solutions of ATRA were prepared in DMSO at10mM and stored at−20◦C wrapped with aluminum foil to avoid direct light irradiation.Dulbecco’s Modified Eagle Medium(DMEM), penicillin–streptomycin and trypsin–EDTA were purchased from Life Technologies Corporation(Grand Island,NY,USA).Sources of the antibodies are as follows:GFAP (sc-33673),CDK2(sc-163),CDK4(sc260)and-actin(sc-1616)were from Santa Cruz Biotechnology(Santa Cruz Biotechnology Inc.,CA);Cyclin D1(#2922),p27 Kip1(#2552),Phospho-Akt(Ser473,#9271),Akt(#9272),-catenin(#9562)and Phospho--catenin(Ser33/37/Thr41,#9561)were from Cell Signaling Technology (Beverly,MA);GSK3(BS1402)and p-GSK3(S9,BS4084)were from Bioworld Technology Inc.(St.Louis,MN,USA).IRDyeTM800conjugated secondary antibodies were obtained from Rockland Inc.(Bedford,PA).2.2.Cell cultureRat C6and human U251glioma cell lines were obtained from the American Type Culture Collection(Manassas,VA,USA),and routinely cultured in DMEM containing 10%fetal bovine serum,1%penicillin and streptomycin,maintained in a humidified atmosphere of95%air,5%CO2at37◦C.Cells were plated in6-well plates or plastic incubatingflask and then treated with the various concentrations of wogonin and ATRA.2.3.Proliferation assayC6and U251cells were seeded into6-well plates at a density of5×105per well and were treated as indicated.Cells were collected after trypsin digestion and counted with a cell counting chamber(Qiujing,Shanghai,China)and micro-scope(YS100;Nikon,Ibaraki,Japan).For cell viability assay,cells were plated into 96-well plates at approximately8000/well in100l medium.Various compounds were added to the cells at indicated concentrations and durations.Cell viability was measured using an MTT assay following the manufacturer’s protocol.2.4.Clonogenic assayCells were plated in35-mm dishes at5000cells per well in DMEM culture medium.Plates were further incubated in incubator for14days until colonies were large enough to be visualized.Colonies were stained with0.01%Crystal violet for 1h and counted under inverted microscope.Experiments were done in triplicate.2.5.Cell cycle analysisCells were starved in serum-free medium for24h to be synchronized,and after treatment with wogonin,cells were harvested and resuspended with70%ethanol overnight at4◦C.Cells were spun down and incubated with the DNA-binding dye propidium iodide(PI)solution[0.1%sodium citrate(w/v),0.1%TritonX-100(v/v), and50mg/L PI in deionized water]for30min at room temperature.Finally,cells were analyzed by FACSCaliburflow cytometry(Becton Dickinson,San Jose,CA). 2.6.Immunofluorescent stainingCells werefixed with4%paraformaldehyde–PBS for30min,washed,and blocked with PBS containing1%BSA for1h at37◦C.The primary antibodies were diluted at certain concentration according to instructions.After being incubated with primary antibody for2h at37◦C,cells were treated with the secondary anti-body at appropriate dilution in1%BSA for1h at37◦C.Nuclei were stained with DAPI (1g/ml).Immunofluorescence photomicrographs were captured usingfluorescent microscope(Olympus IX51,Olympus Corporation,Tokyo,Japan).2.7.Western blot assayCells were collected,washed twice with PBS and lysed in extraction buffer (50mM Tris–HCl pH7.4,150mM NaCl,1mM PMSF,1mM EDTA,1%Triton X-100 and0.1%SDS,200mM b-mercaptoethanol,1mM phenylmethylsulfonylfluoride and 1g/ml aprotinin)for30min on ice.The lysates were centrifuged at13,000×g for 15min,and protein concentration in the supernatants was quantified by BCA assay. Proteins were separated by10%SDS–polyacrylamide gel electrophoresis and elec-troblotted onto nitrocellulose membranes by standard procedures.After blocking with1%BSA in PBS for1h at37◦C,the membranes were incubated for2h with pri-mary antibodies at37◦C followed by IRDye800conjugated secondary antibody for214Y.Wang et al./Toxicology Letters222 (2013) 212–2231h at37◦C.Immunoreactive protein bands were detected with an Odyssey Scanning System(LI-COR Inc.,Lincoln,NE).2.8.Real-time quantitative PCRTotal RNA were extracted from C6and U251cells using TRIzol®Reagent(Invi-trogen,Carlsbad,CA)and then the quality of total RNA was assessed by the ratio of A260/A280using a BioPhotometer(Eppendorf,Hamburg,Germany).1g of total RNA was reverse-transcribed using PrimeScript®RT Reagent Kit with gDNA Eraser(Perfect Real Time).Quantitative real-time PCR(q-PCR)was conducted fol-lowing the protocol supplied by TaKaRa SYBR Green Master Mix kit.All the PCR kits were purchased from TaKaRa Biotechnology Co.,Ltd.(Dalian,China).The rel-ative amount of target mRNA was determined using the comparative threshold (Ct)method by normalizing target mRNA Ct values to those for GAPDH( Ct).The primer sequences are as follows:rat GFAP,forward:5 -ACCTCGGCACCCTGAGGCAG-3 ,reverse:5 -CCAGCGACTCAACCTTCCTC-3 ,and human GFAP,forward:5 -CTGC-GGCTCGATCAACTCA-3 ,reverse:5 -TCCAGCGACTCAATCTTCCTC-3 ;rat GAPDH,for-ward:5 -GTTACCAGGGCTGCCTTCTC-3 ,reverse:5 -GGGTTTCCCGTTGATGACC-3 , and human GAPDH,forward:5 -TGGGTGTGAACCATGAGAAG-3 ,reverse:5 -GCTAAGCAGTTGGTGGTGC-3 .2.9.Rat intracranial glioma modelsTo investigate whether wogonin facilitates the differentiation of C6cells in vivo as well as in cultured cells,C6cells treated with25M wogonin,10M ATRA or not for7days were transplanted into the adult rat brain.For identification within the host tissue,C6cells were labeled with afluorescence dye,DiI-C18-(3),followed by exposure to wogonin for2h prior to transplantation.Before transplantation,C6 cells were pretreated,and then collected,suspended in phosphate buffered saline, complemented with2%of glutamine and1%of penicillin streptomycin.Sprague-Dawley rats weighing from150to180g were group-housed in a temperature-controlled room with a12h:12h light–dark cycle.Animal treatment and maintenance were carried out following the directions in the ethics approval from the Experimentation Ethics Review Committee of China Pharmaceutical Uni-versity.Rats(n=24)were accommodated for one week before experiment and then were randomly divided into three groups(control,wogonin and ATRA)with intracranial injection of different treatment conditions cells.After anesthetized with equithensin(2mL/kg i.p.),animals were placed in the stereotaxic device.A scalp inci-sion was made along the midline and a high-speed drill was used to create a0.5mm diameter burr hole in the skull.For each rat,a10l cell suspension(1×106C6cells) was injected over10min at a depth of6.0mm into the right striatum(3.0mm lat-eral to midline,0.5mm posterior to bregma)using automatic microinjector.5min after the injection,the needle was slowly removed,and the burr hole wasfilled with bone wax.The wound was sutured together,and the rats were allowed to recover. All rats were examined each day.Dead animals were studied,their brains removed and kept in frozen pipes at−80◦C.After20days,the remaining animals were killed. The brains were sectioned and prepared for histology and immunohistochemistry.Then,for study the anti-glioma activity of wogonin in vivo,C6cells were stereo-tactically injected into the right striatum of rats following the above method.Three days after tumor transplantation,animals were randomly divided intofive groups: saline control group,cisplatin(DDP)positive control group,30mg/kg wogonin group,60mg/kg wogonin group and120mg/kg wogonin group.After injecting C6 cells for3days,rats were treated by i.v.with saline,5mg/kg DDP,30mg/kg,60mg/kg and120mg/kg wogonin,respectively,every other day and continued for20days. Tumor volume was estimated following gadolinium enhanced T1-weighted MRI(GE Medical Systems)on day8,13and18.Number of voxels in each slice(slice thickness 1mm,total18slices)were counted using Image J software.Volume=Total number of voxels×0.015625mm3.The brains were excised and thenfixed in4%PFA–PBS for immunofluorescence.2.10.General morphology and immunohistochemistryBrain sections were cut using a cryostat(Leica CM3050S)at15m and collected on glass slides coated with silane and poly-l-lysine.The sections werefixed in4% paraformaldehyde solution and then stored at−80◦C before processing.Microscopic morphology of the brain was assessed by hematoxylin–eosin staining of cryostat sections.The tumor cross sectional areas were measured based on H&E stained cryostat sections from perfusion-fixed brains using computer-assisted image analy-sis as previously described.Tumor sizes were determined according to the following formula:tumor volume=(square root of maximum cross sectional area)3.For immunofluorescence,brain cryostat sections were processed for indirect immunofluorescence.Blocking was performed by incubating60min at room tem-perature with PBS containing5%bovine serum albumin(PBS–BSA).Then,sections were incubated with primary antibodies overnight at4◦C,with3%BSA and0.1% Triton-X.Slides were washed three times in PBS and exposed to FITC-conjugated secondary antibodies for2h at room temperature.Nuclei were counterstained with DAPI.The immunoreactivity wasfinally examined under an Olympus confocal microscope.The PCNA staining was performed using a PCNA cell proliferation detection kit (Keygen,Nanjing,China)according to the manufacturing instructions.Following above-mentioned procedures,the frozen sections were blocked with3%hydrogen peroxide and10%methanol for20min with subsequent rinses with PBS.The spec-imens were incubated with primary antibodies and second antibodies,subsequent treated with the appropriate ABC-complex as chromogenic agent,resulting in brown staining of antigen-expressing cells.2.11.Statistical analysisData were analyzed using post hoc tests included the Student’s t-test and the Tukey multiple comparison tests.All experiments reported here represent at least triplicate independent replications.All data are represented as mean value±SD and the significant difference is indicated as*P<0.05,**P<0.01.3.Results3.1.Wogonin’s effect on the cell viability of glioma C6and U251 cellsTo investigate the cytotoxicity of wogonin on glioma C6and U251cells,we examined cell viability using MTT assay for24h.As shown in Fig.1A and B,wogonin inhibited the growth of C6and U251cells in a concentration-dependent manner.We additionally performed cell count assay showing that the effects of low con-centration of wogonin(25M,50M)on cell growth over longer period of time showed significant proliferation inhibition(Fig.1C and D).Approximately50%growth inhibition was observed when C6and U251cells were exposed to50M wogonin for72h.This is comparable to the results observed with10M ATRA,a known differentiation agent for glioma cells.Next,wogonin-induced cell reproductive death was also examined by evaluating the colony-formation ability using clonogenic assay.As data shown in Fig.1E and F,the clone numbers of C6and U251cells treated with50M wogonin were62%and57%,respectively,as much as that in control. Obviously,treatment with wogonin showed significant decrease in cell survival in a dose-dependent manner.3.2.Wogonin induces glioma cells at G1/G0phase arrestWe carried out FACS analysis on C6and U251cells treated with various wogonin.As shown in Fig.2A and B,wogonin treatment (12.5,25,50M)for24h obviously depleted the G2/M population in a dose-dependent manner.To further investigate whether cells re-enter the cell cycle after wogonin treatment was removed for 4,8,12,and24h,we respectively examined the sub-G1groups of C6and U251cells at each time point,which were pretreated with 50M wogonin for24h.As shown in Fig.2D,after wogonin was removed for24h,cells began to re-enter new cell cycle,indicating that the effect of wogonin inducing sub-G1arrest was reversible. Next,we analyzed expression levels of principal cell-cycle regu-latory proteins including cyclin D1,cyclin-dependent kinase2,4 and the cyclin-dependent kinase inhibitors p27.When cells were treated with various concentrations(0–50M)of wogonin for24h, the proteins levels of cyclin D1,CDK4,and CDK2all decreased as the wogonin concentration was raised.In contrast,the50M wogo-nin treatment resulted in an obvious increase in p27protein levels (Fig.2E and F).These data indicate that wogonin induces G1/G0 phases arrest on glioma cancer cells.3.3.Wogonin promotes the glial differentiation of glioma cellsWogonin induced a dramatic change in glioma C6cell morphol-ogy.As shown in Fig.3A,many smaller and spindle-shaped cells with long synaptic-like structure formed after treatment of wogo-nin(25and50M).Immunofluorescence indicated that a dramatic increase in GFAP production in C6and U251cells treated with wogonin(25M)were remarkably similar to those treated withY.Wang et al./Toxicology Letters222 (2013) 212–223215Fig.1.Wogonin inhibits cell growth in glioma cells.Wogonin-induced cell deaths of two different glioma cells are shown in C6(A)and U251(B).Cells were treated within 0–200M wogonin for24h and cell viability was examined by MTT assay.(C and D)The effect of wogonin on the growth of glioma cells compared with all-trans retinoic acid(ATRA).Cells were treated with different concentrations of wogonin or10M ATRA for72h.(E and F)The effect of wogonin(0–50M)on the colony formation ability of glioma cells.Data are presented as means±SD(n=3).Statistical differences compared with the controls are given as*P<0.05,**P<0.01.ATRA(10M),that is known to induce glioma cell differentiation (Fig.3B).Western blot analysis also showed that wogonin signifi-cantly increased GFAP level after treatment of wogonin(25M)for 3days.This increased expression of GFAP was observed with wogo-nin concentrations ranging from0to50M in C6and U251cells (Fig.3C and D).In addition,gene expression level of GFAP was mea-sured by real time-PCR and it was found that the mRNA expression of GFAP increased in the wogonin treatment group as compared to the control(Fig.3E).All these results suggest that wogonin induces the differentiation of glioma cells into mature glial cells.3.4.Wogonin exerts its differentiation activity via the inhibitionof GSK-3ˇ/ˇ-catenin pathway in glioma cellsSince theflavonoid wogonin has been shown to induce decrease in phosphorylated Akt in several types of cells including malignant216Y.Wang et al./Toxicology Letters222 (2013) 212–223Fig.2.Wogonin induces growth arrest at G0/G1phase of cell cycle in glioma cells.(A and B)The effect of wogonin on cell cycle profile(C6(A)or U251(B))was examined byflow cytometry analysis.Cells were treated with wogonin at indicated concentration(0–50M)for24h.The experiments were repeated three times,and the result of a representative experiment was presented.The quantitative data of cell cycle phase are shown in the(C).(D)The recovery effect of glioma cells to re-enter into cell cycle after50M wogonin treatment was removed for4,8,12and24h.(E and F)Western blotting analysis on principal cell-cycle regulatory proteins in C6and U251cells.Cells were incubated with different concentration wogonin(0–50M)for24h.The quantitative data of cell-cycle regulatory protein levels are shown in the(G and H).Y.Wang et al./Toxicology Letters222 (2013) 212–223217Fig.3.Wogonin promotes the differentiation of glioma cells into mature glial cells.Cells were incubated with0,25and50M wogonin for3days.(A)Wogonin promotes morphological transformation of C6cells that indicated cell differentiation.(B)Immunocytochemistry of GFAP proteins expression.C6cells were exposed to25and50M wogonin at indicated concentration for3days.(C)The effect of wogonin compared with ATRA on GFAP protein by western blot analysis.Cells were treated with25M wogonin or10M ATRA for3days.(D)Concentration-dependent effect of wogonin on GFAP protein.Cells were incubated with0,12.5,25,and50M wogonin for3days.(E)The mRNA level of GFAP protein.Cells were incubated with wogonin(0–50M)for24h.GFAP mRNA levels were measured by realtime-PCR.Data(mean±SD)were representative of at least three independent experiments;**P<0.01vs control,*P<0.05vs control.glioma(Huang et al.,2011;Lu et al.,2008;Parajuli et al.,2011),and GSK3,a downstream target molecule of Akt,plays an important role in astrocytic differentiation of human glioblastoma cells(He et al.,2011;Korur et al.,2009;Li et al.,2010a,b),we hypothe-sized that wogonin induced the differentiation in glioma cells was associated with decreased activity of Akt/GSK3signaling. To test this hypothesis,we analyzed both phosphorylated and non-phosphorylated forms of Akt in C6and U251cells treated with indicated doses wogonin or25M wogonin for various lengths of time(Fig.4A and B).Our data indicated that Akt was constitutively phosphorylated in glioma cells,which was significantly inhibited by the wogonin under a dose-and time dependent manner.Wogo-nin caused a more pronounced decrease in phosphorylation of Akt in a time-dependent manner.As proto-oncoproteins-catenin is putative GSK-3substrates for phosphorylation-dependent inactivation,we tested whether wogonin mediated Akt inhibition could lead to inhibition of GSK-3/-catenin signaling activity. Western blot analysis also confirmed up-regulated expression of GSK3and down-regulated expression of-catenin in glioma cells treated by wogonin as time and concentration increased218Y.Wang et al./Toxicology Letters222 (2013) 212–223Fig.4.Akt/GSK-3/-catenin pathway is involved in wogonin induced the differentiation of glioma cells.Wogonin decreased the level of phosphorylated Akt,depend on dose(A)and duration(B).Dose-and time-dependent inhibition of GSK-3and-catenin activities by wogonin was shown in(C)and(D).The ratios of the quantitative data are shown in the under panels.Cells were cultured for24h with various doses of wogonin,or with50M wogonin for indicated time.The Akt,GSK-3and-catenin protein expression and their phosphorylated levels were examined by western blot analysis.Results are expressed as the mean±S.E.M.of three independent experiments.**P<0.01 vs control,*P<0.05vs control.Y.Wang et al./Toxicology Letters222 (2013) 212–223219(Fig.4C and D).In addition,the phosphorylation levels of GSK3and-catenin presented reverse change tendency corresponding to their each protein levels.To further study-catenin pathway regulating the differentia-tion of glioma cells,20mM LiCl and20ng/ml DKK-1were separately added into wogonin treated glioma cells for3days.LiCl,as a known activator of canonical-catenin signaling,can inhibit GSK3activ-ity and consequently induce accumulation of-catenin.Dkk1can bind to LRP5/6co-receptors for Wnt ligands,and inhibit-catenin signaling.We found that C6cells became round and lose the synap-tic sample structure after LiCl treatment,but Dkk1significantly promoted C6cells differentiating into mature glial cells(Fig.5A). The result shown in Fig.5B also illustrated that the expression level of-catenin was increased by LiCl and decreased by DKK-1. Actually,wogonin caused significant inhibition of-catenin pro-tein compared to the separate drug groups.Treatment of C6and U251cells with LiCl in the presence of wogonin(50M)resulted in apparent down-regulation of GFAP compared to treatment with wogonin alone.The expression of GFAP was upregulated after Dkk1 treatment,and this activation by Dkk1was increase by the com-bined with wogonin treatment.These results suggest that blocking the-catenin signaling treated by Dkk1promotes the differen-tiation of glioma cells,while activating this pathway stimulated by LiCl inhibits glioma cells differentiating into mature glial cells. We thus concluded that wogonin suppressed the GSK3/-catenin signaling through attenuating the phosphorylation of Akt,and fur-ther exerted differentiation-inducing and anti-proliferative effect in glioma cancer cells.3.5.Wogonin promotes differentiation of C6glioma cells in vivoGlioma C6cells were pretreated with wogonin for72h before intracranial implantation to determine whether wogonin facilitates the differentiation of C6cells in vivo as well as in cultured cells,andFig.5.Inhibition of-catenin signaling promotes the glial differentiation in glioma cells.Cells were incubated with50M wogonin,20mM LiCL or20ng/ml Dkk1for72h, respectively.(A)Wogonin,LiCl,and DKK-1induced morphological changes of C6cells(a,control;b,LiCl;c,DKK-1;d,wogonin;e,wogonin and LiCl;f,wogonin and Dkk1).(B)-Catenin and GFAP protein levels were examined by western blot analysis.(C)The ratios of the quantitative data are shown in the under panels.Results are expressed as the mean±S.E.M.of three independent experiments.**P<0.01vs control,*P<0.05vs control.。