固相萃取-高效液相色谱法同时测定蔬菜制品中11种合成着色剂
高效液相色谱法测定蔬菜中除虫脲、灭幼脲和杀铃脲残留

高效液相色谱法测定蔬菜中除虫脲、灭幼脲和杀铃脲残留郭爱华;李堃;李敏;王玮;郭蒙京【摘要】建立高效液相色谱测定蔬菜中除虫脲、灭幼脲和杀铃脲残留的检测方法.蔬菜样品用乙腈提取,经DisQuE分散固相萃取试剂盒净化,C18色谱柱(250mm×4.6 mm,5μm)分离,以乙腈-水(68∶32)溶液洗脱,二极管阵列(PDA)检测器检测,外标法定量.在0.02~1.0 mg/L范围内,除虫脲、灭幼脲和杀铃脲的质量浓度与对应的色谱峰面积线性相关,除虫脲和灭幼脲的检出限为0.010 mg/kg,杀铃脲的检出限为0.015 mg/kg.除虫脲、灭幼脲和杀铃脲标准溶液的色谱峰面积的日内相对标准偏差分别为1.03%,1.31%,0.82%(n=6),日间相对标准偏差分别为1.43%,1.56%,1.06%(n=6).加标回收率为88.7%~108.0%.该方法简单、快速、准确,适合蔬菜中除虫脲、灭幼脲和杀铃脲残留的测定.【期刊名称】《化学分析计量》【年(卷),期】2016(025)003【总页数】4页(P57-60)【关键词】蔬菜;除虫脲;灭幼脲;杀铃脲;高效液相色谱法【作者】郭爱华;李堃;李敏;王玮;郭蒙京【作者单位】北京市西城区疾病预防控制中心,北京 100120;北京市西城区疾病预防控制中心,北京 100120;北京市西城区疾病预防控制中心,北京 100120;北京市西城区疾病预防控制中心,北京 100120;北京市西城区疾病预防控制中心,北京100120【正文语种】中文【中图分类】O657.7除虫脲、灭幼脲和杀铃脲属于苯甲酰脲类杀虫剂,是几丁质合成抑制剂,能干扰靶标昆虫体内几丁质合成而导致其死亡,或直接降解昆虫几丁质[1]。
因该类杀虫剂对人畜毒性相对较低,并能有效地防治对有机磷、有机氯有抗性的害虫,因而被用于多种农作物的害虫防治。
由于该类农药对人类存在潜在危险,2014年国家卫生和计生委发布实施的食品安全国家标准《食品中农药最大残留限量》(GB 2763-2014)[2]规定了食品中除虫脲、灭幼脲和杀铃脲的最大残留量分别为20,3,0.1 mg/kg。
固相萃取-高效液相色谱法检测食品中酸性大红GR

固相萃取-高效液相色谱法检测食品中酸性大红GR武传香;魏莉莉;薛霞;卢兰香;丁一;高敏;王骏;刘艳明【期刊名称】《中国食品添加剂》【年(卷),期】2024(35)3【摘要】建立了高效液相色谱法测定多种食品中非法染料酸性大红GR的分析方法,并对样品的提取溶剂、净化方式及色谱条件进行了优化。
样品采用1%氨化甲醇溶液提取,PWAX萃取柱净化浓缩,以10 mmol乙酸铵-甲醇为流动相,经TitankC18色谱柱分离,采用二极管阵列检测器测定。
结果显示,酸性大红GR在0.040〜10.00μg/mL线性关系良好(R2>0.999),检出限和定量限分别为0.025和0.10 mg/kg。
在阴性样品中添加0.10、0.20、1.00 mg/kg三水平加标,平均回收率为80.14%〜102.60%,相对标准偏差为1.08%〜4.29%。
通过与食品补充检验方法比较,验证了本方法测定辣椒制品、水产制品和饮料中酸性大红GR的准确性,同时对其他食品中酸性大红GR进行测定,结果证明该方法灵敏度高、回收率好,为食品安全的监管提供了技术支持。
【总页数】8页(P246-253)【作者】武传香;魏莉莉;薛霞;卢兰香;丁一;高敏;王骏;刘艳明【作者单位】山东省食品药品检验研究院;产业技术基础公共服务平台;国家市场监管重点实验室(肉及肉制品监管技术)【正文语种】中文【中图分类】TS207.3【相关文献】1.固相萃取.超高效液相串联质谱法同时测定食品中红色2G、酸性大红、酸性橙Ⅱ和酸性金黄G2.固相萃取-高效液相色谱法在食品中合成着色剂检测中的应用3.基于改性碳纳米管分散固相萃取吸附剂-高效液相色谱法检测酱菜中3种食品防腐剂4.基于新型磁性固相萃取材料结合高效液相色谱法检测食品中碱性橙Ⅱ和柠檬黄因版权原因,仅展示原文概要,查看原文内容请购买。
食品理化检验考试重点详解

第二章食品样品的采集保存和处理1、食品中无机元素的测定:可分为湿消化法和干灰化法。
2、湿消化法:是在适量的食品样品中,加入氧化性强酸,加热破坏有机物,使得测的无机成分释放出来,形成不挥发的无机化合物,以便进行分析测定。
3、干灰化法:是将食品放在瓷坩埚中,先在电炉上使样品脱离水,炭化,再置于500C-600C 的高温中灼烧灰化。
使样品中的有机物氧化分解成二氧化碳、水和其他气体而挥发,留下的无机物供测定用。
4、扩散法:加入某种试剂使待测物生成气体而被测定,通常在扩散皿中进行。
5、顶空法:常与气相色谱法联用,通常可分为静态和动态分析法。
静态顶空分析法是将样品置于密闭系统中,恒温加热一段时间达平衡后,取出蒸气相用气相色谱法分析样品待测成分的含量。
动态顶空分析法是在样品顶空分离装置中不断通氮气,使其中挥发性成分随氮气逸出,并收集于吸附柱中,经热解析或溶剂解析后进行分析。
6、固相萃取:实际上就是柱色谱分离方法。
在小柱中填充适当的固定相制成固相萃取柱,当样品液通过小柱时,待测成分被吸留,用适当的溶剂洗涤除去样品基体或杂质,然后用一种选择性的溶剂将待组分洗脱,从而达到分离、净化和浓缩的目的。
第三章食品营养成分分析1粗脂肪:用有机溶剂在索氏提取器中直接提取食品中的脂肪时,因少量脂溶性成分与脂肪混合在一起。
故称粗脂肪。
2总脂肪:在用有机溶剂萃取以前,先加酸或碱进行处理,使食品中结合脂肪游离出来再用有机溶剂萃取后所测得的脂肪。
3维生素:维持机体生命活动过程所必须的一类低分子有机化合物。
4膳食纤维:指存在于食物中不能被人体消化的多糖类和木质素的总和。
5粗纤维:表示食物中不能被烯酸、稀碱、有机溶剂所荣解,不能为人体消化利用的物质。
6食品:各种供人食用或饮用的成品或饮料,以及传统是食品又是药品的物品,但是不包括以治疗为目的的药品。
7营养价值:食品中所含的营养素和能量能满足人体营养需要的程度。
8食品中的营养素成分主要有:蛋白质、脂肪、碳水化合物、维生素、矿物质和水。
肉制品中人工合成着色剂的测定
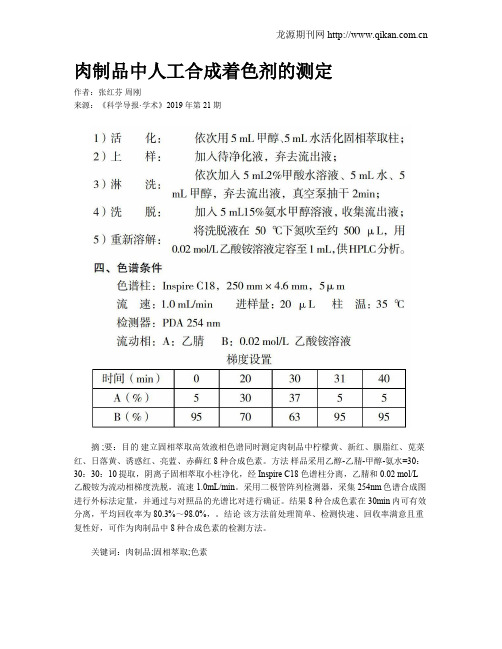
肉制品中人工合成着色剂的测定作者:张红芬周刚来源:《科学导报·学术》2019年第21期摘 ;要:目的建立固相萃取高效液相色谱同时测定肉制品中柠檬黄、新红、胭脂红、苋菜红、日落黄、诱惑红、亮蓝、赤藓红8种合成色素。
方法样品采用乙醇-乙腈-甲醇-氨水=30:30:30:10提取,阴离子固相萃取小柱净化,经Inspire C18色谱柱分离,乙腈和 0.02 mol/L 乙酸铵为流动相梯度洗脱,流速1.0mL/min。
采用二极管阵列检测器,采集254nm色谱合成图进行外标法定量,并通过与对照品的光谱比对进行确证。
结果 8种合成色素在30min内可有效分离,平均回收率为80.3%~98.0%,。
结论该方法前处理简单、检测快速、回收率满意且重复性好,可作为肉制品中8种合成色素的检测方法。
关键词:肉制品;固相萃取;色素食用合成着色剂简称合成色素,为增进人们的食欲,保持食品色泽鲜艳而添加的非营养成分,多以煤焦油中提煉出的苯胺染料为原料研制而成,有含R-N=N-R键、苯环或氧杂蒽等结构[1-3]。
合成色素色泽鲜艳、用量少、性质稳定、易着色,且价格相对便宜,但超标、超范围等不规范使用则会造成人体被动的摄入过量的食用合成色素,导致出现嗳气、偏头痛、多种过敏症状以及中毒等症状,更有甚者造成致癌、致畸、致突变等危害。
目前食品合成色素的检测主要参照国标GB/T5009.35-2003《食品中合成着色的测定》[4]、GB/T5009.141-2003《食品中诱惑红的测定》[5]、SN/T1743-2006《食品中的诱惑红、酸性红、亮蓝、日落黄的含量检测》[6]需采用多个液相系统才能完成对8合成色素的检测,且样品前处理较复杂,检测耗时长。
文献报道[7-8]的这些检测方法多是针对这些人工合成色素中的一种或几种,而没有关于这8种人工合成色素在肉制品中同时检测的报道。
并且这些检测方法的样品净化过程一般都采用聚酰胺吸附法或是液液萃取法(赤藓红)等,这些前处理方法具有操作环境差、操作费时繁琐、回收率低等特点。
食品中9种人工合成着色剂的检测

广东化工2019年第11期·196·第46卷总第397期食品中9种人工合成着色剂的检测吴嘉彦,戴辉(品测(上海)检测科技有限公司,上海201108)The Determination of Nine Synthetic Colors in FoodWu Jiayan,Dai Hui(Pince Testing Co.,Ltd.,Shanghai201108,China)Abstract:This study was based on the national standard method,and use ammonia-70%methanol(1︰99V/V)as extract solvent.Sample solution was purified by weak anion solid phase extraction,and finally carried out by the ultra high pressure liquid chromatography with ultraviolet detector.Analyses were separated by gradient elution and measured by the different waves.Nine types synthetic colors in food that could be measured simultaneously.The limitation of determination was0.2mg/kg,the range of calibration curves was0.2~10mg/L,the range of recoveries was85.0%~110.0%and the repeatability was2.0%~8.9%).Keywards:synthetic colors;colors in food;ultra high pressure liquid chromatography食品着色剂,又称食用色素,是种以给食品着色为主要目的的添加剂。
食品中合成着色剂的测定方法︱食品中合成着色剂的检测分析方法

食品中合成着色剂的测定方法︱食品中合成着色剂的检测分析方法1、主题内容与适用范围本标准规定了食品中合成着色剂的测定方法。
本标准适用于食品中合成着色剂的测定。
第一篇高效液相色谱法(第一法)2、食品中合成着色剂的测定原理食品中人工合成着色剂用聚酰胺吸附法或液-液分配法提取,制成水溶液,注入高效液相色谱仪,经反相色谱别离,根据保存时间定性和与峰面积比拟进行定量。
最小检出量,新红5ng、柠檬黄4ng、苋菜6ng、胭脂红8ng、日落黄7ng、赤藓红18ng、亮蓝26ng,当进样量相当时最低检出浓度分别为;;;;;;。
3、食品中合成着色剂的测定试剂正己烷。
盐酸。
乙酸。
甲醇:经滤膜(0.5?m)过滤。
聚酰胺粉(尼龙6):过200目筛。
乙酸铵溶液(0.02mol/L) :称取乙酸铵,加水至1000mL,溶解,经滤膜(0.45?m)过滤。
氨水:量取氨水2mL,加水至100mL,混匀。
氨水-乙酸铵溶液(0.02mol/L) :量取氨水,加乙酸铵溶(0.02mol/L)至1000mL,混习。
甲醇-甲酸(6+4)溶液:量取甲醇60mL,甲酸40mL,混匀。
柠檬酸溶液:称取20g柠檬酸(C6H8O7·H2O),加水至100mL,溶解混匀。
无水乙醇-氨水-水(7+2+1)溶液:量取无水乙醇70mL、氨20mL、水10mL,混匀。
三正辛胺正丁醇溶液(5%):量取三正辛胺5mL,加正丁醇至100mL,混匀。
饱和硫酸钠溶液。
硫酸钠溶液(2g/L)。
pH6的水:水加柠檬酸溶液调pH值到6。
合成着色剂标准溶液:准确称取按其纯度折算为100%质量的柠檬黄、日落黄、苋菜红、胭脂红、新红、赤藓红、亮蓝、靛蓝各,100mL容量瓶中,加pH6水到刻度。
配成水溶液(1.00mg/mL)。
3.17合成着色剂标准使用液:临用时上述溶液〔或将3.16〕加水稀释20倍,经滤膜〔0.45?m〕过滤。
配成每毫升相当50.0?g的合成着色剂。
4、食品中合成着色剂的测定仪器高效液相色谱仪,带紫外检测器,254nm波长。
矿产

矿产资源开发利用方案编写内容要求及审查大纲
矿产资源开发利用方案编写内容要求及《矿产资源开发利用方案》审查大纲一、概述
㈠矿区位置、隶属关系和企业性质。
如为改扩建矿山, 应说明矿山现状、
特点及存在的主要问题。
㈡编制依据
(1简述项目前期工作进展情况及与有关方面对项目的意向性协议情况。
(2 列出开发利用方案编制所依据的主要基础性资料的名称。
如经储量管理部门认定的矿区地质勘探报告、选矿试验报告、加工利用试验报告、工程地质初评资料、矿区水文资料和供水资料等。
对改、扩建矿山应有生产实际资料, 如矿山总平面现状图、矿床开拓系统图、采场现状图和主要采选设备清单等。
二、矿产品需求现状和预测
㈠该矿产在国内需求情况和市场供应情况
1、矿产品现状及加工利用趋向。
2、国内近、远期的需求量及主要销向预测。
㈡产品价格分析
1、国内矿产品价格现状。
2、矿产品价格稳定性及变化趋势。
三、矿产资源概况
㈠矿区总体概况
1、矿区总体规划情况。
2、矿区矿产资源概况。
3、该设计与矿区总体开发的关系。
㈡该设计项目的资源概况
1、矿床地质及构造特征。
2、矿床开采技术条件及水文地质条件。
高效液相色谱法快速检测食品中12_种添加剂
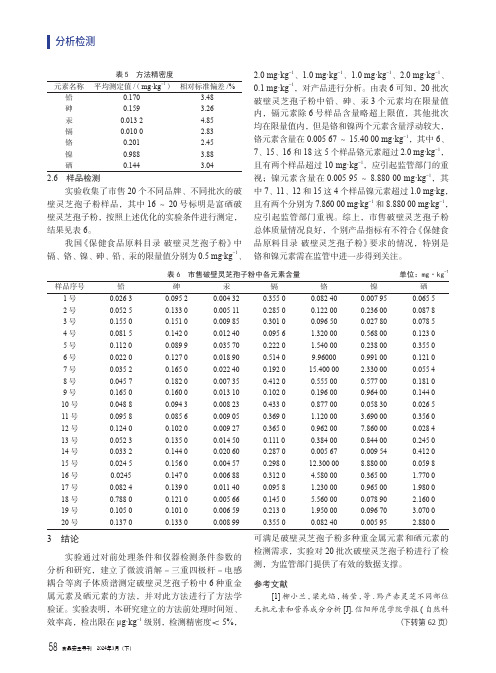
表5 方法精密度元素名称平均测定值/(mg·kg-1)相对标准偏差/%铅0.170 3.48砷0.159 3.26汞0.013 2 4.85镉0.010 0 2.83铬0.201 2.45镍0.988 3.88硒0.144 3.042.6 样品检测实验收集了市售20个不同品牌、不同批次的破壁灵芝孢子粉样品,其中16~20号标明是富硒破壁灵芝孢子粉,按照上述优化的实验条件进行测定,结果见表6。
我国《保健食品原料目录破壁灵芝孢子粉》中镉、铬、镍、砷、铅、汞的限量值分别为0.5 mg·kg-1、2.0 mg·kg-1、1.0 mg·kg-1、1.0 mg·kg-1、2.0 mg·kg-1、0.1 mg·kg-1,对产品进行分析。
由表6可知,20批次破壁灵芝孢子粉中铅、砷、汞3个元素均在限量值内,镉元素除6号样品含量略超上限值,其他批次均在限量值内,但是铬和镍两个元素含量浮动较大,铬元素含量在0.005 67~15.40 00 mg·kg-1,其中6、7、15、16和18这5个样品铬元素超过2.0 mg·kg-1,且有两个样品超过10 mg·kg-1,应引起监管部门的重视;镍元素含量在0.005 95~8.880 00 mg·kg-1,其中7、11、12和15这4个样品镍元素超过1.0 mg·kg,且有两个分别为7.860 00 mg·kg-1和8.880 00 mg·kg-1,应引起监管部门重视。
综上,市售破壁灵芝孢子粉总体质量情况良好,个别产品指标有不符合《保健食品原料目录破壁灵芝孢子粉》要求的情况,特别是铬和镍元素需在监管中进一步得到关注。
表6 市售破壁灵芝孢子粉中各元素含量 单位:mg·kg-1样品序号铅砷汞镉铬镍硒1号0.026 30.095 20.004 320.355 00.082 400.007 950.065 5 2号0.052 50.133 00.005 110.285 00.122 000.236 000.087 8 3号0.155 00.151 00.009 850.301 00.096 500.027 800.078 5 4号0.081 50.142 00.012 400.095 6 1.320 000.568 000.123 0 5号0.112 00.089 90.035 700.222 0 1.540 000.238 000.355 0 6号0.022 00.127 00.018 900.514 09.960000.991 000.121 0 7号0.035 20.165 00.022 400.192 015.400 00 2.330 000.055 4 8号0.045 70.182 00.007 350.412 00.555 000.577 000.181 0 9号0.165 00.160 00.013 100.102 00.196 000.964 000.144 0 10号0.048 80.094 30.008 230.433 00.877 000.058 300.026 5 11号0.095 80.085 60.009 050.369 0 1.120 00 3.690 000.356 0 12号0.124 00.102 00.009 270.365 00.962 007.860 000.028 4 13号0.052 30.135 00.014 500.111 00.384 000.844 000.245 0 14号0.033 20.144 00.020 600.287 00.005 670.009 540.412 0 15号0.024 50.156 00.004 570.298 012.300 008.880 000.059 8 16号0.02450.147 00.006 880.312 0 4.580 000.365 00 1.770 0 17号0.082 40.139 00.011 400.095 8 1.230 000.965 00 1.980 0 18号0.788 00.121 00.005 660.145 0 5.560 000.078 90 2.160 0 19号0.105 00.101 00.006 590.213 0 1.950 000.096 70 3.070 0 20号0.137 00.133 00.008 990.355 00.082 400.005 95 2.880 03 结论实验通过对前处理条件和仪器检测条件参数的分析和研究,建立了微波消解-三重四极杆-电感耦合等离子体质谱测定破壁灵芝孢子粉中6种重金属元素及硒元素的方法,并对此方法进行了方法学验证。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
固相萃取-高效液相色谱法同时测定蔬菜制品中11种合成着色剂刘剑波;余莲芳;朱明扬;高炜【摘要】建立一种固相萃取-高效液相色谱法同时测定蔬菜制品中11种人工合成着色剂的方法.蔬菜制品中的合成着色剂经25%的氨水乙醇提取,弱阴离子固相萃取小柱Waters Oasis WAX净化,Agilent 5 TC-C18(2)为分离柱,甲醇:乙腈(4:1,体积比)-0.02 mol/L乙酸铵溶液为流动相进行梯度洗脱,用二极管阵列检测器多波长检测,外标法定量.结果显示,11种色素在0.2μg/mL~40μg/mL范围内具有良好的线性,在1、2、10 mg/kg 3个加标水平下的平均回收率为87.5%~102.6%,相对标准偏差为1.5%~5.8%,检出限在5μg/L~45μg/L.该方法可用于蔬菜制品中人工合成着色剂的日常检测.【期刊名称】《食品研究与开发》【年(卷),期】2018(039)023【总页数】6页(P141-146)【关键词】固相萃取;高效液相色谱;蔬菜制品;人工合成着色剂【作者】刘剑波;余莲芳;朱明扬;高炜【作者单位】岳阳市食品质量安全监督检验中心,湖南岳阳414000;岳阳市食品质量安全监督检验中心,湖南岳阳414000;岳阳市食品质量安全监督检验中心,湖南岳阳414000;岳阳市食品质量安全监督检验中心,湖南岳阳414000【正文语种】中文着色剂即使食品赋予色泽和改善食品色泽的物质。
食用着色剂分为人工合成着色剂和天然着色剂两大类。
人工合成着色剂由于着色力好,色彩鲜艳,色调多样,成本低廉等原因在食品中被广泛使用[1-2]。
中国允许在食品中添加的人工合成着色剂依据GB2760-2014《食品安全国家标准食品添加剂使用标准》有:柠檬黄、新红、苋菜红、靛蓝、胭脂红、日落黄、诱惑红、亮蓝、酸性红、喹啉黄、赤藓红共11种[3]。
人工合成着色剂种类繁多,人体摄入超过一定量,可引起慢性中毒,致突变性、致畸性、致癌性等风险增加。
蔬菜制品是指以蔬菜和食用菌为原料,采用腌制、干燥、油炸等工艺加工而成的各种产品,包括酱腌菜、蔬菜干制品、食用菌制品、其他蔬菜制品。
蔬菜制品是我国传统的加工方法,在中国居民的膳食结构中,蔬菜制品是深受喜爱的一种佳品。
蔬菜在加工过程中容易退色或变色,因此常常在加工过程中添加食用着色剂以改善其感官色泽[4-7]。
GB2760-2014中规定:人工合成着色剂柠檬黄、苋菜红、靛蓝、胭脂红、亮蓝在腌渍蔬菜中允许使用,其限量分别为:0.1、0.05、0.01、0.05、0.025 g/kg。
而新红、日落黄、酸性红、喹啉黄、诱惑红、赤藓红不得在蔬菜制品中使用[3]。
目前GB 5009.35-2016《食品安全国家标准食品中合成着色剂的测定》中利用聚酰胺吸附法和液-液分配法(适用于含赤藓红的样品)对柠檬黄、新红、苋菜红、靛蓝、胭脂红、日落黄、亮蓝、赤藓红这8种人工合成着色剂进行了检测[8]。
GB 5009.141-2016《食品安全国家标准食品中诱惑红的测定》规定了食品中诱惑红的测定方法[9]。
SN/T 1743-2006《食品中诱惑红、酸性红、亮蓝、日落黄的含量检测》规定了食品中诱惑红,酸性红,亮蓝,日落黄的检测方法[10]。
DBS 32/012-2016《食品安全地方标准食品中喹啉黄的检测》中规定了食品中喹啉黄的检测方法[11]。
要同时检测这11种色素,需要采用4种标准方法。
因此建立一种蔬菜制品中11种人工合成着色剂的高通量检测方法对于实际应用是非常必要的。
本文旨在建立一种利用固相萃取-高效液相色谱法同时测定蔬菜制品中这11种着色剂的方法,简化前处理步骤,优化色谱条件,一次检测,提高日常监督检测的针对性和效率。
1 材料与方法1.1 材料与试剂检测样品为2017年岳阳市食品质量安全监督检验中心从本市内抽取的蔬菜制品如香辣榨菜、剁椒风味包、老坛酸菜包、酸豆角、麻辣海带、油炒萝卜、藕片、莴笋片、香辣金针菇、酸笋风味包、盐渍生姜、芋头条、冬瓜片、南瓜脆片、秋葵脆片、黄瓜片、红薯干、土豆条、原味香菇、脱水胡萝卜。
甲醇、乙腈(色谱纯):美国TEDIA公司;乙酸铵(≥98%):J&K科技;25%氨水、乙醇、柠檬酸、甲酸(分析纯):国药集团化学试剂有限公司;试验用水均为超纯水。
柠檬黄、苋菜红、胭脂红、日落黄、诱惑红、赤藓红、亮蓝标准溶液:中国计量科学研究院;新红标准溶液:农业部环境保护科研监测所;靛蓝(90.0%)、喹啉黄(97.2%):Dr.Ehrenstorfer公司;酸性红(90.7%):曼哈格公司。
1.2 仪器与设备U3000超高效液相色谱仪(配有二极管阵列检测器)、Smart 2pure超纯水机:美国赛默飞公司;H1850R高速冷冻离心机:湖南湘仪仪器有限公司;BAS224S电子分析天平:德国赛多利斯股份公司;RE-2000B旋转蒸发器:上海亚荣生化仪器厂;KQ-600E超声波清洗器:昆山市超声仪器有限公司;涡旋仪QL-866:海门市其林贝尔仪器制造有限公司;Sepline-S4全自动固相萃取仪:莱伯泰科;Oasis WAX固相萃取小柱(150 mg/6 cc):美国 Waters公司。
1.3 方法1.3.1 样品处理准确称取样品1 g至2 g于10 mL离心管中,加入5 mL25%的氨水乙醇,涡旋混匀后超声提取10 min,高速冷冻离心10 min(4℃,10 000 r/min)后立即将上清液转入鸡心瓶中,残渣重复提取2次,合并3次上清液。
40℃减压旋转蒸发至5 mL左右,用水转移入玻璃管中,用200 g/L柠檬酸调pH值至5.0左右,待Oasis WAX小柱净化。
将净化柱装入全自动固相萃取仪,把进样管和收集管放入指定位置,编辑相应的方法管理程序,填充试剂通道并清洗进样针之后,依次按照方法用6 mL甲醇和6 mL纯水润洗小柱,上样时过柱的速度设置为1.0 mL/min,不收集滤液。
再用甲醇∶甲酸∶水=2∶1∶7(体积比)的溶液10 mL淋洗小柱,干燥时间设置为10 s。
最后用15 mL10%氨化甲醇洗脱,收集洗脱液,干燥时间设置为15 s。
取出洗脱液于50℃氮吹至近干后,用2 mL 20%甲醇水溶液溶解残渣,过0.45 μm聚四氟乙烯(poly tetra fluoroethylene,PTFE)滤膜,待测。
1.3.2 标准溶液的制备配制0.2 μg/mL~40 μg/mL 浓度范围的柠檬黄、新红、苋菜红、靛蓝、胭脂红、日落黄、诱惑红、亮蓝、酸性红、喹啉黄、赤藓红的混合标准溶液。
现用现配。
喹啉黄主要由4个峰组成(分别为QYNa2Ⅰ、QYNa2Ⅱ、QYNaⅠ、QYNaⅡ),本方法把4个峰设为一组,由软件进行组校准。
1.3.3 色谱条件色谱柱:Agilent 5 TC-C18(2)250 mm×4.6 mm;柱温30℃;流动相A:甲醇∶乙腈(4∶1),流动相B:0.02 mol/L乙酸铵;流速1 mL/min;液相色谱流动相梯度洗脱程序见表1,多通道检测,柠檬黄、喹啉黄415 nm,新红、苋菜红、胭脂红、日落黄、诱惑红、酸性红、赤藓红515 nm,靛蓝、亮蓝610 nm。
表1 液相色谱流动相梯度洗脱程序Table 1 Gradient elution program of liquid chromatography时间/min 流动相A/% 流动相B/%0 15 85 6 25 75 9 45 5512 80 20 16 80 20 18 15 852 结果与分析2.1 前处理条件的优化2.1.1 样品提取本文研究的蔬菜制品对象主要分为腌制蔬菜、脱水蔬菜和膨化蔬菜三大类。
通过参考文献以及国标方法主要考察甲醇-水溶液、乙醇-氨水-水溶液以及氨水-乙醇溶液对样品中色素的提取效果,甲醇-水溶液的提取液浑浊,不利于转移上清液以及过柱,而膨化蔬菜具有一定的油脂,脱水蔬菜吸水性较强,所以不宜采用水比例较大的提取液。
其中氨水-乙醇作为提取溶剂,样品中的色素提取较为完全。
通过试验发现随着氨水-乙醇溶液中氨水浓度的提高,合成色素的溶出更加充分,但提取液却越浑浊,最后确定用25%的氨水-乙醇作为提取溶剂。
超声后进行高速冷冻离心,然后立即转移上清液能更好地分离油脂和水层[12-16]。
2.1.2 样品净化本文研究的11种有机合成着色剂都含有磺酸基团,因此选择弱阴离子固相萃取小柱Waters Oasis WAX(150 mg/6 cc)对样品进行净化处理。
提取液浓缩转移后使用柠檬酸调pH值至5.0左右,可以使合成色素的磺酸基成电离状态,有利于提高色素在净化柱上的保留性和选择性。
试验中使用含有一定甲酸的甲醇水溶液作为淋洗液既能有效地洗掉天然色素等杂质又能取得较满意的回收率。
通过试验比较了氨水乙醇、氨水甲醇以及甲醇作为洗脱液时在净化柱上的回收率,氨化甲醇的使用结果较为满意。
氨化甲醇溶液氨水所占的比例越高,洗脱能力越强,但不利于氮吹浓缩,最终确定氨化甲醇的浓度为10%。
国标方法中含赤藓红的样品需要用液-液分配法单独处理,本文由于赤藓红在Oasis WAX柱上的保留性较强,所以加大洗脱液用量至15 mL。
样品在最后定容时使用20%甲醇水溶液溶解残渣,有效地保证了赤藓红的回收率。
过滤样品溶液时应选择吸附作用小的聚四氟乙烯(PTFE)滤膜[12-16]。
2.2 色谱条件的优化2.2.1 流动相检测时使用的流动相既要能够使目标物得到有效的分离,又要能改善峰形,提高灵敏度。
本文选用甲醇∶乙腈(4∶1)作为流动相A,0.02 mol/L乙酸铵作为流动相B。
乙腈的加入能显著地改善峰的不对称性,提高灵敏度。
而在流动相中作为调节剂的无机电解质的加入对于改善可电离物质的分离以及获得一个合理且较短的分析时间是很有必要的。
乙酸铵缓冲溶液的浓度定为0.02 mol/L,乙酸铵的浓度越大,由于盐析效应增强导致分析物与色谱柱固定相的作用增强,因此分析物的保留时间增加[16]。
乙酸铵缓冲溶液的浓度为0.02 mol/L时,溶液pH值约为6.5,所有着色剂都以中性化合物存在,以便发生反相机制,因此缓冲溶液的pH值无需优化。
2.2.2 梯度洗脱程序本文使用4 μg/mL的11种合成着色剂的混合标准溶液来试验最佳的分离条件。
最初9 min内有机相比例缓慢上升,使柠檬黄、新红、苋菜红、靛蓝达到有效分离。
9 min至12 min,加快了有机相上升的速率,分离胭脂红、日落黄、喹啉黄,然后高有机相比例保持几分钟使诱惑红、酸性红、赤藓红、亮蓝依次洗脱下来,最后2 min回到初始流动相比例来平衡色谱柱。
优化的梯度程序见表1。
2.2.3 检测波长本试验采用二极管阵列检测器在190 nm~800 nm范围内进行全波长扫描,找到各个组分的特征吸收波长。