BORONIC ACIDS AS LIGANDS FOR AFFINITY CHROMATOGRAPHY
USP
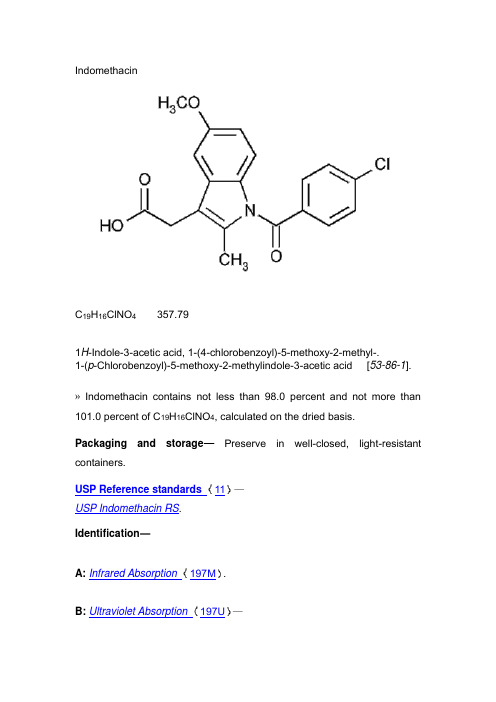
IndomethacinC19H16ClNO4357.791H-Indole-3-acetic acid, 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-.1-(p-Chlorobenzoyl)-5-methoxy-2-methylindole-3-acetic acid [53-86-1].»Indomethacin contains not less than 98.0 percent and not more than 101.0 percent of C19H16ClNO4, calculated on the dried basis.Packaging and storage—Preserve in well-closed, light-resistant containers.USP Reference standards 11—USP Indomethacin RS.Identification—A: Infrared Absorption 197M.B: Ultraviolet Absorption 197U—Solution: 25 µg per mL.Medium: hydrochloric acid in methanol (1 in 120).Absorptivities at 318 nm, calculated on the dried basis, do not differ by more than 3.0%.C: Its X-ray diffraction pattern (see X-ray Diffraction 941) conforms to that of USP Indomethacin RS.Loss on drying 731— Dry it at a pressure below 5 mm of mercury at100for 2 hours: it loses not more than 0.5% of its weight.Residue on ignition 281: not more than 0.2%.Heavy metals, Method II 231: 0.002%.Assay—Mobile phase— Prepare a suitable solution of 0.01 M monobasic sodium phosphate and 0.01 M dibasic sodium phosphate in acetonitrile and water (approximately 1:1).Standard preparation—Dissolve an accurately weighed quantity of USP Indomethacin RS in Mobile phase to obtain a solution having a known concentration of about 0.1 mg per mL.Assay preparation— Weigh accurately about 100 mg of Indomethacin, and transfer to a 100-mL volumetric flask. Dissolve in and dilute with Mobile phase to volume, and mix. Pipet 10 mL of this solution into a 100-mL volumetric flask, dilute with Mobile phase to volume, and mix.Chromatographic system(see Chromatography 621)—The liquidchromatograph is equipped with a 254-nm detector and a 4-mm × 30-cmcolumn that contains 10-µm packing L1. The flow rate is about 1 mL perminute. Chromatograph the Standard preparation, and record the peakresponses as directed under Procedure:the column efficiency determinedfrom the analyte peak is not less than 500 theoretical plates, and therelative standard deviation for replicate injections is not more than 1.0%.Procedure— Separately inject equal volumes (about 20 µL) of the Standardpreparation and the Assay preparation into the chromatograph, record thechromatograms, and measure the responses for the major peaks. Calculatethe quantity, in mg, of C19H16ClNO4 in the portion of Indomethacin taken bythe formula:1000C(r U / r S)in which C is the concentration, in mg per mL, of USP Indomethacin RS inthe Standard preparation; and r U and r S are the peak responses obtained atequivalent retention times from the Assay preparation and the Standardpreparation, respectively.Auxiliary Information—Please check for your question in the FAQsbefore contacting USP.estion Contact Expert Committeeph Clydewyn M. Anthony, Ph.D.Scientist1-301-816-8139(MDCCA05) Monograph Development-Cough Cold andAnalgesicse s Lili Wang, Technical Services Scientist1-301-816-8129RSTech@USP32–NF27 Page 2632Chromatographic Column—INDOMETHACINChromatographic columns text is not derived from, and not part of, USP 32 or NF 27.消炎痛C19H16ClNO4357.791H-Indole-3-acetic酸、1 -(4-chlorobenzoyl)5-methoxy-2-methyl -。
药学 专业文献 翻译

药学专业文献翻译Enzymatic Synthesis and Antioxidant Properties of Poly (rutin) 聚(芦丁)的酶促合成与抗氧化性质†‡†‡††Motoichi Kurisawa, Joo Eun Chung, Hiroshi Uyama,* and Shiro Kobayashi*Abstract 摘要Rutin, quercetin-3-rutinoside, is one of the most famous glycosides of flavonoid and widely present in many plants. In this study, we performed an oxidative polymerization of rutin using Myceliophthora laccase as catalyst in a mixture of methanol and buffer to produce a flavonoid polymer and evaluated antioxidant properties of the resultant polymer. Under selected conditions , the polymer with molecular weight of several thousands was obtained in good yields. The resulting polymer was readily soluble in water, DMF, and DMSO, although rutin monomer showed very low water solubility. UV measurement showed that the polymer had broad transition peaks around 255 and 350 nm in water, which were red-shifted in an alkaline solution. Electron spin resonance (ESR) measurement showed the presence of a radical in the polymer. The polymer showed greatly improved superoxide scavenging activity and inhibition effects on human low-density lipoprotein (LDL) oxidation initiated by 2,2‘-azobis(2-amidino-propane) dihydrochloride (AAPH), compared with the rutin monomer. The polymer also protected endothelial cells from oxidative injuryinduced by AAPH as a radical generator with a much greater effect than the rutin monomer.芦丁,槲皮素芸香糖苷,是最著名的黄酮类糖苷之一,广泛存在于很多植物中。
苏沃雷生的合成

z
a
t
i
on,andt
hepu
r
i
t
s
yi
99.
89%.Thes
t
r
uc
t
u
r
eo
ft
het
a
r
tc
ompoundi
sc
on
f
i
rmedby1H-NMRand13C
-NMR.Theop
t
imi
z
edr
ou
t
eha
sadvan
t
age
so
f
ge
mi
l
dr
e
a
c
t
i
onc
ond
i
t
i
onsands
2020
02
27;修回日期:
2020
03
30;责任编辑:张士莹
基金项目:河北省自然科学基金(
B2019208137)
第一作者简介:杨媛媛(
1994—),女,河北沧州人,硕士研究生,主要从事药物化学方面的研究。
通讯作者:张
勇教授。E-ma
i
l:
zhangyong@hebus
t.
edu.
cn
杨媛媛,张志光,李明乐,等 .
唑2
-基)苯甲酰基]
1,
4
-二氮环庚烷1
-基]
1,
3
-苯并[
d]恶唑(简称化合物 1,下同),由美国默沙东公司研发,
[]
2014 年经 FDA 批准上市,是首个经批准上市的双食欲素受体拮抗剂,主要用于治疗原发性失眠症 5 。临床
纳米钯_乙二醇体系催化Suzuki反应的研究

·94·
广州化工
2009年 37卷第 6期
2 结果与讨论
211 碱对 Suzuk i反应的影响
碱对偶联反应的影响是比较明显的 。因此我们以对甲氧基 溴苯和苯硼酸作为基准反应 ,以不同的碱进行 Suzuki反应 ,从中 选择最合适的碱试剂 。我们发现 ,该体系下 , K2 CO3、NaOH、NaH2 CO3 的效果都比较好 。其他的碱试剂 ,如 K3 PO4 ·3H2O、Na3 PO4 ·12H2O、Na2 HPO4 ·12H2O ,相对产率都较低 。相比之下 ,我们 选择相对较廉价的 K2 CO3 作为该体系的碱试剂 。
表 1 不同碱对 Suzuki反应的影响
碱
产率 / %
Na2 CO3
63
N aHCO 3
81
K2 CO3
84
KF·2H2O
39
KOH
48
N aOH
83
K3 PO4 ·3H2O
77
Na3 PO4 ·12H2O
73
Na2 HPO4 ·12H2O
72
212 温度对 Suzuk i反应的影响
考察了温度对 Suzuki反应影响 。以对甲氧基溴苯和苯硼酸 作为底物作为模型反应 。实验结果见表 2。
表 2 不同温度对 Suzuki反应产率的影响
温度 / ℃
产率 / %
60
72
70
77
80
84
90
83
100
85
实验结果 表 明 , 产 率 随 着 温 度 上 升 有 小 幅 度 上 升 。达 到 80℃之后 ,再继续升温对产率则几乎没有什么影响 。反应温度 为 80℃为宜 。
213 催化剂用量对 Suzuk i反应的影响
RSC Adv-2015

a
College of Material Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou, 310036, China. E-mail: jinhua6903@; Fax: +86-57128866903; Tel: +86-571-28866903 Institute of Analytical and Applied Chemistry, Department of Chemistry, Zhejiang University, Hangzhou, 310027, China Qianjiang College, Hangzhou Normal University, Hangzhou, 310036, China information (ESI) available. See DOI:
RSC Advances
PAPER Thiol-functionalized silica microspheres for online preconcentration and determination of mercury species in seawater by high performance liquid chromatography and inductively coupled plasma mass spectrometry†
1. Introduction
Mercury has become a global environmental concern, especially in the form of methylmercury (MeHg), by virtue of global transport and the biogeochemical cycle.1 The toxicity and bioavailability of mercury are species-specic. It was reported that organomercuric compounds are generally more toxic than inorganic mercuric species and elemental mercury.2 The earth's oceans supply human beings with hundreds and thousands of different kinds of seafood. Considering their high bioaccumulation and biomagnication in the food chain, the amounts of mercury species in seawater are vital to the quality of seafood. Besides, mercury speciation analysis in seawater is benecial to further understand the biogeochemical cycling of mercury.2 The development of accurate and sensitive analytical
氨基酸的结晶

Crystallization of Amino Acids on Self-AssembledMonolayers of Rigid Thiols on GoldAlfred Y.Lee,†,‡Abraham Ulman,†,§and Allan S.Myerson*,‡Department of Chemical and Environmental Engineering,Illinois Institute of Technology, Chicago,Illinois60616,and Department of Chemical Engineering,Chemistry and Material Science,and the NSF MRSEC for Polymers at Engineered Interfaces,Polytechnic University,Brooklyn,New York11201Received March6,2002.In Final Form:May3,2002Self-assembled monolayers(SAMs)of rigid biphenyl thiols are employed as heterogeneous nucleants for the crystallization of L-alanine and DL-valine.Powder X-ray diffraction and interfacial angle measurements reveal that the L-alanine crystallographic planes corresponding to nucleation are{200}, {020},and{011}on SAMs of4′-hydroxy-(4-mercaptobiphenyl),4′-methyl-(4-mercaptobiphenyl),and4-(4-mercaptophenyl)pyridine on gold(111)surfaces,respectively.In the case of DL-valine,monolayer surfaces that act as hydrogen bond acceptors(e.g.,4′-hydroxy-(4-mercaptobiphenyl)and4-(4-mercaptophenyl)-pyridine)induce the racemic crystal to nucleate from the{020}plane whereas the nucleating plane for the4′-methyl-(4-mercaptobiphenyl)surface is the fast-growing{100}face.The observation of crystal nucleation and orientation can be attributed to the strong interfacial interactions,in particular,hydrogen bonding,between the surface functionalities of the monolayer film and the individual molecules of the crystallizing phase.Molecular modeling studies are also undertaken to examine the molecular recognition process across the interface between the surfactant monolayer and the crystallographic planes.Similar to binding studies of solvents and impurities on crystal habit surfaces,binding energies between SAMs and particular amino acid crystal faces are calculated and the results are in good agreement with the observed nucleation planes of the amino acids.In addition to L-alanine and DL-valine,the interaction of SAMs and mixed SAMs of rigid thiols on the morphology of R-glycine is examined(Kang,J.F.;Zaccaro, J.;Ulman,A.;Myerson,ngmuir2000,16,3791),and similarly the calculations are in good agreement. These results suggest that binding energy calculations can be a valid method to screen self-assembled monolayers as potential templates for nucleation and growth of organic and inorganic crystals.I.IntroductionCrystallization from solution is a two-step process: nucleation,the birth of a crystal,and crystal growth,the growth of the crystal to larger sizes.2In this process, prenucleation aggregates(or clusters)are formed by individual molecules,which become stable nuclei,upon reaching a critical size,and further grow into macroscopic crystals.Homogeneous nucleation is very rare and re-quires high supersaturation to surmount the activation barrier,∆G crit.However,for a fixed supersaturation the activation barrier can be lowered by decreasing the surface energy of the aggregate,for instance,by introducing a foreign surface or substance.3This foreign surface(or substance)includes“tailor-made”additives,4impurities,5 organic single crystals,6Langmuir monolayers7floating at the air-water interface,and self-assembled monolayers (SAMs)immersed in solution.1Tailor-made additives or auxiliaries are designer impurities that have one part which resembles the crystallizing species and another part that is chemically or structurally different from the solute molecule.4,8These additives disrupt the bonding sequence in the crystals,thereby lowering the growth rate of the affected faces as evident in the case of L-alanine where hydrophobic amino acids such as L-leucine and L-valine inhibited the development of specific crystal faces,while in the presence of hydrophilic amino acids the crystal morphology did not change.9In addition to being habit modifiers,these molecular additives can also control polymorphism,where the impurities inhibit the growth of one polymorph and,in turn,promote the growth of the other polymorph.10Nucleation promoters such as organic single crystals and self-assembled monolayers have also been used to control polymorph selectivity,based on geometric match-ing between the molecular clusters and the ledges of the crystal substrates11and interfacial hydrogen bonding between the monolayer film and solute clusters,12respec-*To whom correspondence should be addressed.Phone:312 5677010.Fax:3125677018.E-mail:myerson@.†Polytechnic University.‡Illinois Institute of Technology.§NSF MRSEC for Polymers at Engineered Interfaces.(1)Kang,J.F.;Zaccaro,J.;Ulman,A.;Myerson,ngmuir2000, 16,3791.(2)(a)Myerson,A.S.Handbook of Industrial Crystallization,2nd ed.;Butterworth-Heinemann:Boston,2002.(b)Myerson,A.S.Mo-lecular Modeling Applications in Crystallization;Cambridge Uni-versity Press:New York,1999.(c)Mullin,J.W.Crystallization,4th ed.;Butterworth-Heinemann:Boston,2001.(3)Turnbull,D.J.Chem.Phys.1949,18,198.(b)Fletcher,N.H.J. Chem.Phys.1963,38,237.(4)Weissbuch,I.;Lahav,M.;Leiserowitz,L.In Molecular Modeling Applications in Crystallization;Myerson, A.S.,Ed.;Cambridge University Press:New York,1999;p166.(5)Meenan,P.A.;Anderson,S.R.;Klug,D.L.In Handbook of Industrial Crystallization,2nd ed.;Myerson,A.S.,Ed.;Butterworth Heinemann:Boston,2002;p67.(6)Carter,P.W.;Ward,M.D.J.Am.Chem.Soc.1993,115,11521.(7)(a)Rapaport,H.;Kuzmenko,I.;Berfeld,M.;Kjaer,K.;Als-Nielsen, J.;Popovitz-Biro,R.;Weissbuch,I.;Lahav,M.;Leiserowitz,L.J.Phys. Chem.B2000,104,1399.(b)Frostman,L.M.;Ward,ngmuir 1997,13,330.(8)(a)Berkovitch-Yellin,Z.;Ariel,S.;Leiserowitz,L.J.Am.Chem. Soc.1985,105,765.(b)Addadi,L.;Weinstein,S.;Gate,E.;Weissbuch,I.;Lahav,M.J.Am.Chem.Soc.1982,104,4610.(9)Li,L.;Lechuga-Ballesteros,D.;Szkudlarek,B.A.;Rodriguez-Hornedo,N.J.Colloid Interface Sci.1994,168,8.(10)(a)Weissbuch,I.;Lahav,M.;Leiserowitz,L.Adv.Mater.1994, 6,952.(b)Davey,R.J.;Blagden,N.;Potts,G.D.;Docherty,R.J.Am. Chem.Soc.1997,119,1767.5886Langmuir2002,18,5886-589810.1021/la025704w CCC:$22.00©2002American Chemical SocietyPublished on Web06/22/2002tively.Similar to Langmuir monolayers,self-assembled monolayers can be used as an interface across which stereochemical matching13and hydrogen bonding14in-teraction can transfer order and symmetry from the monolayer surface to a growing crystal.However,SAMs and mixed SAMs15lack the mobility of molecules at an air-water interface and hence the possibility to adjust lateral positions to match a face of a nucleating crystal. This is clearly evident in the case of the SAMs of rigid biphenyl thiols,where even conformational adjustment is not possible.Recently,SAMs of4-mercaptobiphenyl have been shown to be more superior to those of al-kanethiolates and are stable model surfaces.16Further-more,the ability to engineer surface functionalities at the molecular level makes SAMs of rigid thiols very attractiveas templates for heterogeneous nucleation. Organosilane monolayer films have been used to promote nucleation and growth of calcium oxalate mono-hydrate crystals17and have been employed in“biomimetic”synthesis as observed in the oriented growth of CaCO318 and iron hydroxide crystals.19Functionalized SAMs of alkanethiols have also been shown to control the oriented growth of CaCO3.20This was also evident in the hetero-geneous nucleation and growth of malonic acid crystals21 on alkanethiolate SAMs on gold where the monolayer composition strongly influenced the orientation of the malonic acid crystals.Additionally,functionalized alkane-thiolate SAMs have enhanced the growth of protein crystals.22More recently,SAMs and mixed SAMs of rigid thiols served as templates.1It was observed that glycine nucleated in the R-form independent of the hydroxyl and pyridine surface concentration and the morphology of the glycine crystal was very sensitive to the OH and pyridine site densities.Self-assembled monolayers on solid surfaces offer many advantages for enhanced crystal nucleation.In this work, SAMs of rigid thiols on gold are employed to investigate the effects of interfacial molecular recognition on nucle-ation and growth of L-alanine and DL-valine crystals.In addition,molecular modeling techniques are employed to examine the affinity between monolayer surfaces and particular amino acid crystal faces and to gain a better understanding of the molecular recognition events oc-curring.The modeling techniques employed are similar to studies of solvent and additive interactions on crystal habit23but have never been applied to organic monolayer films as templates for nucleation.II.Experimental SectionMaterials.Anhydrous ethanol was obtained from Pharmco (Brookfield,CT).L-Alanine(CH3CH(NH2)CO2H),and DL-valine ((CH3)2CHCH(NH2)CO2H)were purchased from Aldrich and used without further purification.Distilled water purified with a Milli-Q water system(Millipore)was used.Details of the synthesis of the4′-substituted4-mercaptobiphenyl(see Figure1)are described elsewhere.24Gold Substrate and Monolayer Preparation.Glass slides were cleaned in ethanol in an ultrasonic bath at40°C for10min. The slides were next treated in a plasma chamber at an argon pressure of0.1Torr for30min.Afterward,they were mounted in the vacuum evaporator(Key High Vacuum)on a substrate holder,approximately15cm above the gold cluster.The slides were baked overnight in a vacuum(10-7Torr)at300°C.Gold (purity>99.99%)was evaporated at a rate of3-5Å/s until the film thickness reached1000Å;the evaporation rate and film thickness were monitored with a quartz crystal microbalance (TM100model from Maxtek Inc.).The gold substrates were annealed in a vacuum at300°C for18h.After cooling to room temperature,the chamber was filled with high-purity nitrogen and the gold slides were either placed into the adsorbing solution right after the ellipsometric measurement was performed or stored in a vacuum desiccator for later use.25Atomic force microscopy(AFM)studies24revealed terraces of Au(111)with typical crystalline sizes of0.5-1µm2.Monolayers were formed by overnight(∼18h)immersion of clean substrates in10µm ethanol solutions of the thiols.The substrates were removed from the solution,rinsed with copious amounts of absolute ethanol to remove unbound thiols,and blown dry with a jet of nitrogen. Contact angle measurements,IR spectroscopy,and ellipsometry showed that after1h,90%or more of the SAMs are formed.26 Thus,to ensure equilibrium SAMs,the gold substrates were left overnight in the dipping solution.Crystal Growth.Nucleation and growth experiments were carried out in Quartex jars(1oz.)at25°C.Supersaturated solutions(25%)of L-alanine and DL-valine were obtained by dissolving 4.58g and 1.95g in22.0g of Millipore water, respectively.The solutions were heated to65°C for90min in an ultrasonic bath to obtain complete dissolution.The solutions were cooled to room temperature for90min before the SAMs were carefully introduced and aligned vertically to the wall. Macrocrystals of L-alanine and DL-valine nucleated at the surfaces(11)(a)Bonafede,S.J.;Ward,M.D.J.Am.Chem.Soc.1995,117, 7853.(b)Mitchell,C.A.;Yu,L.;Ward,M.D.J.Am.Chem.Soc.2001, 123,10830.(12)Carter,P.W.;Ward,M.D.J.Am.Chem.Soc.1994,116,769.(13)(a)Landau,E.M.;Levanon,M.;Leiserowitz,L.;Lahav,M.;Sagiv, J.Nature1985,318,353.(b)Weissbuch,I.;Berfeld,M.;Bouwman,W.; Kjaer,K.;Als,J.;Lahav,M.;Leiserowitz,L.J.Am.Chem.Soc.1997, 119,933.(14)Weissbuch,I.;Popvitz,R.;Lahav,M.;Leiserowitz,L.Acta Crystallogr.1995,B51,115.(15)For a review on SAMs of thiols on gold see:(a)Ulman,A.An Introduction to Ultrathin Organic Films:From Langmuir-Blodgett to Self-Assembly;Academic Press:Boston,1991.(b)Ulman,A.Chem. Rev.1996,96,1533.(16)(a)Kang,J.F.;Ulman,A.;Liao,S.;Jordan,R.J.Am.Chem.Soc. 1998,120,9662.(b)Kang,J.F.;Jordan,R.;Ulman,ngmuir1998,14,3983.(17)Campbell,A.A.;Fryxell,G.E.;Graff,G.L.;Rieke,P.C.; Tarasevich,B.J.Scanning Microsc.1993,7(1),423.(18)Archibald,D.D.;Qadri,S.B.;Gaber,ngmuir1996,12, 538.(19)Tarasevich,B.J.;Rieke,P.C.;Liu,J.Chem.Mater.1996,8,292.(20)Aizenberg,J.;Black,A.J.;Whitesides,G.M.J.Am.Chem.Soc. 1999,121,4500.(21)Frostman,L.M.;Bader,M.M.;Ward,ngmuir1994, 10,576.(22)Ji,D.;Arnold,C.M.;Graupe,M.;Beadle,E.;Dunn,R.V.;Phan, M.N.;Villazana,R.J.;Benson,R.;Colorado,R.,Jr.;Lee,T.R.;Friedman, J.M.J.Cryst.Growth2000,218,390.(23)(a)Docherty,R.;Meenan,P.In Molecular Modeling Applications in Crystallization;Myerson,A.S.,Ed.;Cambridge University Press: New York,1999;p106.(b)Myerson,A.S.;Jang,S.M.J.Cryst.Growth 1995,156,459.(c)Walker,E.M.;Roberts,K.J.;Maginn,ngmuir 1998,14,5620.(d)Evans,J.;Lee,A.Y.;Myerson,A.S.In Crystallization and Solidification Properties of Lipids;Widlak,N.,Hartel,R.W.,Narine, S.,Eds.;AOCS Press:Champaign,IL,2001;p17.(24)Kang,J.F.;Ulman,A.;Liao,S.;Jordan,R.;Yang,G.;Liu,G. Langmuir2001,17,95.(25)(a)Jordan,R.;Ulman,A.J.Am.Chem.Soc.1998,120,243.(b) Jordan,R.;Ulman,A.;Kang,J.F.;Rafailovich,M.;Sokolov,J.J.Am. Chem.Soc.1999,121,1016.(26)Ulman,A.Acc.Chem.Res.2001,34,855.Figure1.Rigid4′-substituted4-mercaptobiphenyls.Crystallization of Amino Acids on Thiol SAMs Langmuir,Vol.18,No.15,20025887and near the edge of the substrates.Only crystals having visible SAM area around them were considered,and the rest were discarded.The chosen crystals attached to the substrates were removed from the solution and stored in a vacuum desiccator for later analysis.Due to the strong adhesion of the crystal face to the SAM surface,gold marks were often observed on the crystal face that nucleated on the SAM surface.Characterization.A Rudolph Research AutoEL ellipsometer was used to measure the thickness of the monolayer surface.The He -Ne laser (632.8nm)light fell at 70°on the sample and reflected into the analyzer.Data were taken over five to seven spots on each sample.The measured thickness of the SAMs of biphenyl thiols ranged from 12to 14Å,assuming a refractive index of 1.462for all films.Powder X-ray diffraction patterns of crystalline L -alanine and DL -valine were obtained with a Rigaku Miniflex diffractometer with Cu K R radiation (λ)1.5418Å).All samples were manually ground into fine powder and packed in glass slides for analysis.Data were collected from 5°to 50°with a step size of 0.1°.Crystal habits of L -alanine and DL -valine were indexed by measuring the interfacial angles using a two-circle optical goniometer.All possible measured interfacial angles were compared with the theoretical values derived from the unit cell parameters of L -alanine and DL -valine crystals.27,28III.Modeling SectionIII.1.General.All of the binding energy calculations,including molecular mechanics and dynamics simulations,are carried out with the program Cerius 2.The overall methodology and procedures are summarized in Figure 2.The crystal structures of each amino acid are obtained from the Cambridge Crystallographic Database (ref codes GLYCIN17,LALNIN12,and VALIDL for R -glycine,L -alanine,and DL -valine,respectively).To accurately predict the crystal morphology,molecular mechanics simulations using a suitable potential function (or force field)are performed.In this work,molecular simulations are carried out using the DREIDING 2.21force field.29The van der Waals forces are approximated with the Lennard-Jones 12-6expression,and hydrogen bonding energy is modeled using a Lennard-Jones-like 12-10expression.The Ewald summation technique is employed for the summation of long-range van der Waals and electrostatic interactions under the periodic boundary conditions,and the charge distribution within the molecule is calculated using the Gasteiger method.30ttice Energy Calculation.The lattice energy E lat,also known as the cohesive or crystal binding energy,is calculated by summing all the atom -atom interactions between a central molecule and all the surrounding molecules in the crystal.If the central molecule and the n surrounding molecules each have n ′atoms,thenwhere V kij is the interaction between atom i in the central molecule and atom j in the k th surrounding parison to the “experimental”lattice energy,V exp ,allows us to assess the accuracy of the intermolecular interactions between the molecules by the defined po-tential function.where the term 2RT represents a compensation factor for the difference between the vibrational contribution to the crystal enthalpy and gas-phase enthalpy 31and ∆H sub is the experimental sublimation energy.III.3.Morphological Predictions.The morphology of each amino acid crystal is predicted using the attach-ment energy (AE)32calculation and the Bravais -Friedel -Donnay -Harker (BFDH)law.33The habit or shape of the crystal depends on the growth rate of the faces present.Faces that are slow growing have the greatest morpho-logical importance,and conversely,faces that are fast growing have the least morphological importance and are the smallest faces on the grown crystal.The simplest morphological simulation is the BFDH law which assumes that the linear growth rate of a given crystal face is inversely proportional to the corresponding interplanar distance after taking into account the extinction conditions of the crystal space group.The attachment energy of a crystal face is the difference between the crystal energy and the slice energy.Hartman and Bennema 32found that the relative growth rate of a face is directly proportional to the attachment energy and as a result,the more negative the attachment energy (or more energy released)for a particular face,the less prominent that face is on the crystal.Conversely,faces with the lowest attachment energies are the slowest growing faces and thus have the greatest morphological importance .III.4.Molecular Modeling of SAMs of 4-Mercapto-biphenyls on a Au(111)Surface.Molecular dynamics (MD)simulations are useful techniques in gaining insights on the structural and dynamical properties of self-assembled monolayers.In contrast to molecular mechan-ics,molecular dynamics computes the forces and moves the atom in response to forces,while molecular mechanics computes the forces on the atoms and changes their position to minimize the interaction energy.Recently,MD simulations have been used to investigate the packing order and orientation of rigid 4-mercaptobiphenyl thiol monolayers on gold surfaces.Results show that hydrogen-terminated biphenylmercaptan packs in the herringbone conformation 34and suggest average tilt angles of 8°.(27)Simpson,H.J.;Marsh,R.E.Acta Crystallogr .1966,20,550.(28)Mallikarjunan,M.;Rao,S.T.Acta Crystallogr .1969,B25,296.(29)Mayo,S.L.;Olafson,B.D.;Goddard,W.A.,III J.Phys.Chem .1990,94,8897.(30)Gasteiger,J.;Marsili,M.Tetrahedron 1980,36,3219.(31)Williams,D.E.J.Phys.Chem .1966,45,3370.(32)(a)Hartman,P.;Bennema,P.J.Cryst.Growth 1980,49,145.(33)(a)Bravais,A.Etudes Crystallographiques ;Gauthier-Villars:Paris,1866.(b)Friedel,M.G.Bulletin de la Societe Francaise de Mineralogie 1907,30,326.(c)Donnay,J.D.;Harker,D.Am.Mineral.1937,22,446.Figure 2.Overall scheme showing the computational meth-odology adopted when calculating the binding energy betweenthe crystallographic plane and the monolayer surface.Elat)∑k )1n ∑i )1n ′∑j )1n ′V kij (1)V exp )-∆H sub -2RT(2)5888Langmuir,Vol.18,No.15,2002Lee et al.Based on this work,molecular mechanics simulations are performed for hydroxy-and methyl-terminated 4-mer-captobiphenyl along with 4-(4-mercaptophenyl)pyridine for binding studies with different crystallographic planes.In the periodic model,each unit cell contains four biphenyl molecules and the geometric parameters are a )10.02Å,b )42.25Å,c )10.11Åand R )138.3°, )119.9°,γ)95.7°.The length in the y -direction is set to ∼42Åto ensure two-dimensional periodicity.Also,the gold atoms are arranged in a hexagonal lattice along the XY plane with a nearest neighbor atom of 2.88Å,and the biphenyl occupied a ( 3× 3)R30°Au(111)lattice.To simulate different 4′-substituted 4-mercaptobiphenyls,minimiza-tion was carried out by fixing the biphenyl moiety and varying the substituents at the 4′-position.As a result,the simulated models yielded uniform ordered SAMs of 4′-substituted 4-mercaptobiphenyls and 4-(4-mercapto-phenyl)pyridine with identical packing structure and dynamics to those of a hydrogen-terminated monolayer of biphenylmercaptan (Figure 3).However,this is not true experimentally since adsorption of different 4′-substituted 4-mercaptobiphenyls on gold surfaces results in different monolayer structures and thus one of the main assump-tions made in this work.III.5.Binding of Crystal Habit Faces to SAMs of 4-Mercaptobiphenyls on a Au(111)Surface.Based on BFDH and attachment energy morphology prediction,crystal habit faces with the highest morphological im-portance are chosen for binding studies.The crystal surfaces of interest are cleaved and extended to a 3×3unit cell and partially fixed,allowing flexibility in the tail atoms of the amino acid molecules and a more accurate representation of the effects of SAMs of rigid thiols on the crystallographic plane in the calculation of binding energies.The crystal surface is then docked onto a 3×1×3partially fixed nonperiodic monolayer surface,and the conjugate gradient energy minimization technique is performed.Next,the crystal surface is moved to another site on the monolayer surface and the minimization calculations are again performed.This process was repeated 15-20times to obtain the global minimum.For each monolayer surface,numerous calculations are carried out with different crystallographic planes of each amino acid.The binding energy (φBE )of each crystallographic surface with the monolayer surface iswhere φIE is the minimum interaction energy of the monolayer and crystal surfaces,φM is the minimum energy of the monolayer surface in the absence of the crystal face but in the same conformation as it adopts on the surface,and φS is the minimum energy of the crystal surface with no monolayer surface present and in the same molecular conformation in which it docks on the surface.Negative values of binding energies indicate preferential binding of the crystallographic surfaces with SAMs of 4-mercaptobiphenyl.In cases where the binding energy is positive,there is a less likely chance that the particular crystal face will interact and nucleate on the monolayer surface.Thus,using this approach it is possible to screen self-assembled monolayers as possible templates for nucleation and growth of crystals.IV.Results and DiscussionIV.1.Crystallization of Amino Acids on SAMs on Gold.L -Alanine crystallizes from water in the ortho-rhombic space group P 21212(a )6.025Å,b )12.324Å,and c )5.783Å),27and the morphology of the crystals is bipyramidal,dominated by the {020},{120},{110},and {011}growth forms,35as shown in Figure 4.The crystal grown in aqueous solution is indexed by comparing the interfacial angles measured by optical goniometry and theoretical values based on the unit cell of L -alanine.Powder X-ray diffraction patterns (Figure 5)and inter-facial angle measurements reveal that L -alanine crystals nucleating on SAM surfaces crystallize in the ortho-rhombic space group with similar unit cell dimensions.However,functionalized SAMs induce the formation of L -alanine crystals in different crystallographic directions.L -Alanine crystals display the normal bipyramidal habit but are randomly oriented with the different surfaces.In methyl-terminated SAMs,L -alanine selectively nucle-ated on the {020}plane on the surface (Figure 6),whereas in 100%OH SAM surfaces,L -alanine nucleated on an unobserved {200}side face.The crystal exhibits a similar morphology as observed in aqueous solution with an appearance of a {200}face adjacent to the {110}planes (Figure 6).In both cases,the area of each crystal face is substantially larger than those of the other faces on the crystal.The SAM surfaces almost act as an additive or impurity molecule specifically interacting with the crystal face and consequently reducing the relative growth rate and modifying the habit.Crystallization of L -alanine on 4-(4-mercaptophenyl)pyridine surfaces resulted in the {011}face as the plane corresponding to nucleation (Figure 6).The preferential interaction of the monolayer with the {011}face can be attributed to hydrogen bonding at the crystal -monolayer interface.Unlike the other two sur-faces where they can serve as both hydrogen bond donors and acceptors (4′-hydroxy-4-mercaptobiphenyl)or solely as H-bond donors (4′-methyl-4-mercaptobiphenyl),the pyridine electron pair at the surface only serve as hydrogen bond acceptors.The binding of the pyridine surface and the {011}plane can be explained by the amino and methyl groups protruding out perpendicular to the plane (Figure 7)and forming N -H ‚‚‚N and C -H ‚‚‚N hydrogen bonds with the SAM surface,respectively.In contrast,the 100%(34)Ulman,A.;Kang,J.F.;Shnidman,Y.;Liao,S.;Jordan,R.;Choi,G.Y.;Zaccaro,J.;Myerson,A.S.;Rafailovich,M.;Sokolov,J.;Flesicher,C.Rev.Mol.Biotech .2000,74,175.(35)Lehmann,M.S.;Koetzle,T.F.;Hamilton,W.C.J.Am.Chem.Soc .1972.101,2657.Figure 3.Snapshots of (a)4′-methyl-4-mercaptobiphenyl,(b)4′-hydroxy-4-mercaptobiphenyl,(c)4-(4-mercaptophenyl)pyri-dine,and (d)mixed SAMs of 4′-methyl-4-mercaptobiphenyl and 4′-hydroxy-4-mercaptobiphenyl (top view).φBE )φIE -(φM +φS )(3)Crystallization of Amino Acids on Thiol SAMs Langmuir,Vol.18,No.15,20025889CH 3and 100%OH SAM surfaces do not interact as strongly with the hydrogen bond donating plane.In a similar manner,the appearance of an unobserved {200}face of L -alanine grown in aqueous solution on [Au]-S -C 6H 4-C 6H 4-OH can be attributed to hydrogen bonds forming between the two surfaces.The {200}surface contains alternating methyl (CH 3)and carboxylic groups (COO -)that form N -H ‚‚‚O and O ‚‚‚H -O with the hydroxide group of the monolayer film (Figure 7),ideal for binding with surfaces that can serve as both hydrogen bond donors and acceptors.As a result,the preferential interaction leads to the stabilization and appearance of the {200}face.The oriented nucleation of L -alanine crystals on func-tionalized SAMs arises due to the different molecular structures of each crystal face.Similar to the adsorption of additive onto a crystal face,the interaction (or binding)with the monolayer surface depends on the functional group that each crystal face possesses.As a result of preferential interactions with specific crystal faces,in-terfacial molecular recognition directs nucleation and subsequently influences the crystal growth.In addition to L -alanine,SAMs of rigid thiols are employed to investigate the possibility of inhibiting the racemic crystal and inducing the formation of one of its enantiomers.The powder X-ray diffraction pattern (Figure 8)reveals that DL -valine nucleates in the monoclinic form independent of the hydroxyl,methyl,or pyridine surface concentration and that there was no trace of conglomer-ates.DL -Valine crystallizes in the monoclinic space group P 21/c with a unit cell of dimensions a )5.21Å,b )22.10Å,c )5.41Å,and )109.2°.28Although the structural literature reports three separate space group assignments,Leiserowitz and co-workers 36have shown that two of the three space groups (P 21and P 1)are highly improbable for racemic crystals.Interfacial angle measurements and powder X-ray diffraction undertaken in this work agreed much better with the theoretical values and simulated pattern based on the unit cell of the monoclinic space group(36)Wolf,S.G.;Berkovitch-Yellin,Z.;Lahav,M.;Leiserowitz,L.Mol.Cryst.Liq.Cryst .1990,186,3.Figure 4.Crystallographic image (a)and morphology (b)of L -alanine crystal grown from aqueoussolution.Figure 5.X-ray diffractograms of L -alanine nucleated on functionalized SAMs,compared with L -alanine crystallized from aqueous solution (bottom).Indices of the crystallographic planes corresponding to the diffraction intensities of major peaks are indicated at the top.5890Langmuir,Vol.18,No.15,2002Lee et al.。
Ultrasound-promoted intramolecular direct arylation in a capillary flow microreactor
Ultrasound-promoted intramolecular direct arylation in a capillary flow microreactorLei Zhang,Mei Geng,Peng Teng,Dan Zhao,Xi Lu,Jian-Xin Li ⇑State Key Lab of Analytical Chemistry for Life Science,School of Chemistry and Chemical Engineering,Nanjing University,Nanjing 210093,PR Chinaa r t i c l e i n f o Article history:Received 23March 2011Accepted 20July 2011Available online 26July 2011Keywords:Direct arylationUltrasound irradiation MicroreactorPalladium-catalyzed Synthesisa b s t r a c tAn intramolecular direct arylation of various aryl bromides was performed using ultrasonic irradiation and a continuous flow capillary microreactor.The present procedure provided a higher functional group tolerance,ligand-free,milder reaction conditions and a shorter reaction time for the direct arylation com-pared with the conventional methods.The ultrasonic irritation not only greatly promoted the conversion and selectivity of the direct arylation,but also solved the clogging problem of the microreactor for solid-forming reaction and made the reaction run smoothly.Ó2011Elsevier B.V.All rights reserved.1.IntroductionBiaryl structural motif is observed in a variety of compounds,from natural bioactive products such as vancomycin and schizan-drin [1,2]to some important synthetic compounds like BINOL and BINAP [3,4].An arsenal of well-know catalytic methods includ-ing the Suzuki–Miyaura,Negishi and Stille couplings has been developed to build aryl-aryl bonds [5],however,all these important reactions require the preparation of pre-activated orga-nometallic reagents such as boronic acids,organo-magnesium,-tin or -zinc derivatives that always associated with additional reaction steps,wastes,solvents,purifications and time.Direct arylation as a greener and effective alternative to these commonly employed cross-coupling reactions has attracted considerable interest in re-cent years [6,7],in which the aryl–heteroaryl or aryl–aryl bonds formation of heteroaromatic compounds and simple electron-rich arenes such as pyrroles,indoles,thiophenes and phenols with aryl halides has significantly been developed [6,8–10].However,in spite of their advantages,these reactions usually need complex catalyst systems (complex ligands)and additives (e.g.silver salts),or often suffer from drawbacks such as longer reaction times,high-er temperatures (120–150°C)that significantly limit their scope and functional group tolerance [11–13].So there is still a need for general protocols on the construction of aryl–aryl bonds with a high yield,excellent selectivity and high functional group toler-ance under mild reaction conditions.Microreactor technology has received a great deal of attention for years.It has been shown to afford numerous advantages over traditional stirred-batch reactors,such as increased surface-to-vol-ume ratios,excellent mass-and heat-transfer capabilities,better reaction yields and so on,all of which would be expected to promote highly effective chemical reactions [14–16].However,one main drawback of microreactors is a clogging problem of the small reactor channels when solid-forming reactions are per-formed [17].The easy solution to this problem is highly desirable.It is well known that ultrasound irradiation has been recognized as an efficient technique in organic synthesis to accelerate the reac-tion,improve yield,shorten reaction time and increase selectivity [18–20].As part of our continuing work directed toward developing rea-sonable reaction conditions for aryl–aryl bond formation that are environmentally benign and efficient [21],a methodology study on palladium-catalyzed intramolecular direct arylation reactions using a continuous flow capillary microreactor under ultrasound irradiation was performed.Herein,we report a ligand-free and effi-cient procedure for the intramolecular direct arylation of aryl bro-mides under ultrasonic irradiation and mild conditions in the capillary microreactor.The current reactions possessed a broad substrate scope and high functional group tolerance in good to excellent yields.Furthermore,ultrasonic irradiation solved the clogging problem in microreactor.The microreactor was assem-bled simply from syringes,a conventional capillary,a mixing unit,syringe pumps and a ultrasonic cleaner,all of which are cheap and commercially available (Fig.1).2.Results and discussionIn order to obtain appropriate reaction conditions,an optimi-zation of the reaction conditions was conducted using a batch1350-4177/$-see front matter Ó2011Elsevier B.V.All rights reserved.doi:10.1016/j.ultsonch.2011.07.008Corresponding author.Tel./fax:+862583686419.E-mail address:lijxnju@ (J.-X.Li).reactor.It is reported that N,N-dimethylacetamide (DMA)is supe-rior to other solvents such as N,N-dimethylformamide (DMF),tol-uene,dioxanes and acetonitrile for direct arylation reactions [11,13,22],therefore,as an initial screen,reactions of 2-iodoben-zyl phenyl ether (1)as a substrate with 5mol%Pd(OAc)2and two equiv.of different bases were performed under nitrogen atmo-sphere at 100°C for 24h (Table 1).In pure DMA and inorganic base such as K 2CO 3,Cs 2CO 3and KOAc,substrate 1provided intra-molecular coupling product 20a ,however,both conversions and selectivity (ratio of 20a versus deiodination product 20b )were poor.While organic bases such as 4-dimethylamiopryidine (DMAP)and 1,4-diazabicyclo[2.2.2]octane (DABCO)were ineffec-tive.When a co-solvents (DMA:water)was employed for the reactions,the conversion was greatly increased.For the bases,K 2CO 3and Cs 2CO 3gave an improved conversion,while,NaOAc gave an inferior result.Meaningfully,in the present of KOAc and a 10:1mixture of DMA and water,the conversion was in-creased up to 90%,however,the reaction selectivity was almost unchanged (Table 1,entry 1vs.6,2vs.7,and 5vs.9).Fortu-nately,with changing the substrate 1to 2-bromobenzyl phenyl ether (2),the selectivity was improved more than twice,from 14:1to 32:1(Table 1,entry 9and 10),however,the conversion became to be in a moderate level.In order to optimize the ratio of DMA and water,the effects ofresulted in a lower conversion.It might be reasoned that by per-forming the reaction in the presence of water,the solubility of the inorganic components was increased,but too much water re-duced the solubility of the organic components.The present 10:1ratio might be the optimal point to balance the solubilities of both organic and inorganic components.In all co-solvent reactions,0.05equiv.of tetrabutylammonium bromide (TBAB)was added in order to prevent the generation of palladium black and make the cata-lytic system more stable [23].Based on the above findings,a continuous flow capillary mic-roreactor was employed under the optimal conditions obtained from the batch reaction.When taking 2-iodobenzyl phenyl ether (1)to run the intramolecular direct coupling in the microreactor at 90°C,as time went by the precipitate formed during the reac-tion was adsorbed on the wall of capillary and increasingly accu-mulated,and finally the microreactor was clogged.Due to the short reaction time caused by clogging issue (less than 2h),the reaction in the microreactor only gave a 8.7%conversion and the selectivity was quite lower (7:1).As sonication enables the rapid dispersion of solids and benefits reactions which are the effects arise from cavitation [18,24],this gave us a clue to exposing the capillary coil to ultrasound irradiation,and might solve clogging problem of the microreactor.Fortunately,as ultrasound irradiation was applied,the blockage in the microreactor was easily removed Fig.1.Microreactor system.Entry Substrate Base Solvent Conversion (%)20a :20b 11K 2CO 3DMA 51.516:121Cs 2CO 3DMA 54.418:131DMAP DMA Trace –41DABCO DMA Trace –51KOAc DMA66.115:16b 1K 2CO 3DMA/H 2O (10:1)77.715:17b 1Cs 2CO 3DMA/H 2O (10:1)72.118:18b 1NaOAc DMA/H 2O (10:1)58.88:19b 1KOAc DMA/H 2O (10:1)90.114:110b 2KOAc DMA/H 2O (10:1)70.332:111b3KOAcDMA/H 2O(10:1)Trace–Reaction conditions:2-halobenzyl phenyl ether (0.5mmol),Pd(OAc)2and base were dissolved in each solvent (5.0mL)and heated to 100°C under nitrogen atmosphere for 24h.aDetermined by 1H NMR.b5mol%TBAB was added.obtained when 2-iodobenzyl phenyl ether 1as starting material run for 6h at 90°C (Table 2,entry 3),while batch reaction afforded 90%conversion for 24h at 100°C.However,the reaction selectivity improved very little by comparison with that of batch reaction.It is reported that for the intramolecular direct arylation,despite the fact that aryl iodides provide greater reactivity in cross-coupling reactions,aryl bromides afford better outcomes than aryl iodides [17],thus,a bromide of the substrate (2)was employed for the reaction to improve both conversion and selectivity.As expected,compared with batch reaction,the selectivity and conversion im-proved dramatically from 32:1to 44:1and 70%to 97%,respectively (Table 1,entry 10vs.Table 2,entry 6).While,batch reaction under irradiation afforded 56%conversion and 32:1selectivity.The re-sults confirmed that a combination use of microreactor and ultra-sound greatly benefited the reaction.With this optimal reaction conditions,the scope of the current procedure was examined.Taking the reaction superiority and cheap price into account,aryl bromides were used as substrates.The efficiency and functional tolerance of this procedure have been fully demonstrated by synthesizing a number of functionalized coupling products which bearing substitutes such as nitro,alde-hyde,ketone,ester,phenyl,methyl,methoxy,as well as protecting group tosyl and Bco.The results were outlined in Table 3.Interest-ingly,the electronic nature of the substituents seemed to have lit-tle effect on the product yields.Meaningfully,chloro substituents ether tether could be replaced by amine and amide,and afford the same high yields (entry 14,15).Poly fused N-heterocycles could be prepared in moderate to good results (entry 11-13and 17).Five membered O-heterocycles was also obtained in 97%yield (entry 18).Moreover,a high regioselectivity was observed in cases where direct arylation occurred at two chemically different arene positions,in this case,arylation arose at the more sterically acces-sible position (entry 2and 4).With meta-nitro and -t -butyl substi-tutes,only one product was observed from 1H NMR (entry2and 4).It is noteworthy that the quinolone core was successfully embed-ded in coupling product 38(entry 17)and the current reaction pro-vided a novel way to prepare the quinolone derivatives that possess various bioactivities [26].Conventional intramolecular direct arylation usually needed a higher temperature (above 120o C),complex ligands,additives (e.g.expensive silver salts or pivalic acid),inert atmosphere and a long reaction time (more than 8h)[11–13,22,27],these condi-tions were greatly meliorated in the current reaction procedure.3.ConclusionIn conclusion,we have described a mild and efficient intramo-lecular direct arylation of aryl bromides in a continuous flow cap-illary microreactor with assistance of ultrasonic irradiation.The intramolecular direct arylation was achieved with high selectivity,ligand-free,low temperature,broad substrate scope and compati-ble to complex chemical structures such as quinolone core.Fur-thermore,ultrasound irradiation not only greatly improved the reaction conversion and selectivity,but also solved the clogging problem of microreactor for solid-forming reactions and made the reaction run smoothly.Further research on aryl chloride intra-molecilar coupling and other solid-forming reactions in the mic-roreactor under ultrasound irradiation is underway in our lab.4.Experimental4.1.Reagents and apparatusAll reagents and solvents were used as supplied without further purification.For the batch reaction,all the experiments were .X RT Conversion (%)1X =I 268.918:12X =I 471.620:13X =I 698.4(89.3)17:14X =Br 241.942:15X =Br 467.650:16X =Br 696.7(92.1)44:17X =Cl 6Trace –8b X =Br 644.820:19cX =Br656.134:1RT:reaction time (hour).Conversion was determined by 1H NMR and the isolated yield is shown in parentheses.aDetermined by 1HNMR.bBatch reaction without ultrasound.cUltrasound-assisted batch reaction.Fig.2.Effects of the ratio of DMA to water on the conversion and selectivity.19(2012)250–256L.Zhang et al./Ultrasonics Sonochemistry19(2012)250–256253Table3Biaryl formation in microreactor under ultrasound irradiation.(continued on next page)Scientific Equipment Co.,Ltd.;syringe was SGE supplied by Beijing Huadr Science and Technology Co.,Ltd.;capillary was purchased from Yongnian Ruipu Chromatogram Equipment Co.,Ltd.;1H NMR and 13C NMR were determined in CDCl 3on a Brucker 300or 500MHz spectrometer at room temperature,tetramethylsilane (TMS)served as an internal standard.EI-MS was taken on a QP2010GC–MS instrument (Shimadzu,Japan),ESI-MS and high resolution MS (HRMS)were carried out on a LTQ Orbitrap XL (The Thermo Scientific,USA).Sonication was performed in Kun-shan SB-3200DT ultrasonic cleaner with the frequency of 40kHz and an output power of 150W.4.2.General procedure for the synthesis of substrates 1–14,15,17,and 19(Table 3,entry1–8,12,14,15,16)The appropriate phenol or heterocycle compounds or sulfamide (1.2equiv.),K 2CO 3(or KOH for 12,13,14,and 15)(2equiv.)and DMF (or DMSO for 12,13,14,and 15)(0.5M)were placed in a round bottom flask equipped with a magnetic stir bar.Addition of the appropriate 2-halobenzylbromide (or 4-nitroflorobenzene for 19)(1equiv.)was followed by heating (60°C)of the reaction overnight.The reaction was allowed to cool and was diluted with water and extracted with Et 2O.The combined organic phase was washed by brine,dried over anhydrous Na 2SO 4,filtered and the volatiles were evaporated under reduced pressure.The residues were then purified via silica gel column chromatography usingethyl acetate/hexanes mixtures.Products were characterized by 1H NMR and MS (EI or ESI).4.3.Typical procedure for the synthesis of substrate 16(Table 3,entry 13)To a mixture of 2-halobenzoic acid (1equiv.),DCC (1.2equiv.),DMAP (0.1equiv.)and dichloromethane (0.25M)was added a solution of monomethylaniline (1.5equiv.)in dichloromethane (0.25M)in ice-water bath.When reaction complete,the mixture was filtered and washed with ether.Remove solvent under re-duced pressure.The residues were then purified via silica gel col-umn chromatography using ethyl acetate/hexanes mixtures to give substrate 16.Products were characterized by 1H NMR and EI-MS.4.4.General procedure for intramolecular direct arylation in the microreactor with assistance of ultrasonic irradiationA stock solution of the 2-halobenzyl phenyl ether (0.5mmol)and TBAB (0.05equiv.)in DMA (2.5mL)was prepared and taken up in a SGE gas-tight syringe.A second stock solution containing Pd(OAc)2(5mol%)and KOAc (2.0equiv.)in H 2O (0.45mL)and DMA (2.05mL)was also prepared and taken up in a second SGE gas-tight syringe.The syringes were placed on a TS2-60syringe pump which was set at different flow rates to meet differentTable 3(continued )Entry SubstrateRT Product Isolate yield 13395.914390.615696.016691.317376.618697.3RT:Reaction time (hour).254L.Zhang et al./Ultrasonics Sonochemistry 19(2012)250–256reaction times(Reaction time was calculated according to the fol-lowing equation:time(h)Âflow rate(l L/h)=volume of microre-actor(l L)).The two reactant streams were mixed in a1:1ratio through a micro static mixing Tee,and then delivered into a 500cm capillary coil with an inner diameter of530l m(Fig.1). The capillary coil was located in the maximum energy area in the ultrasonic generator and heated to90°C.The output from the reactor was quenched with Et2O/H2O immediately.The result-ing mixture was extracted with Et2O or EtOAc(3Â8mL)and the combined organic phase was washed with brine,dried over anhy-drous Na2SO4,and then the solvent was removed under reduced pressure.The residues were then purified via silica gel column chromatography using ethyl acetate/hexanes mixtures.Products were characterized by NMR,MS and HRMS(EI or ESI).4.5.Experimental data for compounds4.5.1.Substrates1–19The structures of the known compounds1–3,5–7,9,10,14–18 and21were identified by comparison the spectral data with those reported in the literatures[11–13,28–33].pound4.White solid;Yield:88.1%;1H NMR(300MHz, CDCl3)d7.62–7.59(m,2H),7.38(m,2H),7.24–7.13(m,2H),6.99–6.88(m,2H),5.20(s,2H),1.44(s,9H);EI-MS m/z(rel.int.,%):318 (12),169(100).pound8.White solid;Yield:96.8%;1H NMR(300MHz, CDCl3)d7.67–7.58(m,2H),7.55(dd,J=7.9,1.2Hz,1H),7.48–7.29 (m,6H),7.29–7.21(m,1H),7.16(dd,J=7.7,1.6Hz,1H),7.13–7.07 (m,1H),7.06–7.03(m,J=9.6,1.2Hz,1H),5.13(s,2H);EI-MS m/z (rel.int.,%):338(33),259,(28),169(100).pound11.White solid;Yield:95.6%;1H NMR (300MHz,CDCl3)d9.85(s,1H),7.59(dd,J=7.9,1.1Hz,1H),7.53 (d,J=7.7Hz,1H),7.45(d,J=1.8Hz,1H),7.41(dd,J=8.2,1.9Hz, 1H),7.33(td,J=7.6,1.1Hz,1H),7.19(td,J=7.8,1.6Hz,1H),6.96 (d,J=8.2Hz,1H), 5.30(s,2H), 3.97(s,3H);EI-MS m/z(rel. int.,%):320(11),169(100).pound12.White solid;Yield:96.0%;1H NMR (300MHz,CDCl3)d7.87(dd,J=7.8, 1.6Hz,1H),7.81(d, J=7.7Hz,1H),7.57(dd,J=8.0,1.0Hz,1H),7.53–7.43(m,1H), 7.38(td,J=7.6,1.0Hz,1H),7.19(td,J=8.0,1.6Hz,1H),7.02(dd, J=11.6, 4.3Hz,2H), 5.22(s,2H), 3.93(s,3H);EI-MS m/z(rel. int.,%):320(3),209(56),169(100).pound13.White solid;Yield:83.9%;1H NMR (300MHz,CDCl3)d8.03–7.85(m,2H),7.60(dd,J=7.9,1.1Hz, 1H),7.52(d,J=7.7Hz,1H),7.34(td,J=7.5,1.1Hz,1H),7.21(td, J=7.8,1.7Hz,1H),7.10–6.95(m,2H),5.20(s,2H),2.56(s,3H); EI-MS m/z(rel.int.,%):304(12),169(100).pound19.White solid;Yield:95.7%;1H NMR (300MHz,CDCl3)d7.60(d,J=7.9Hz,1H),7.53(d,J=7.6Hz,1H), 7.37–7.29(m,2H),7.25–7.15(m,2H),6.98–6.91(m,2H),5.15(s, 6H),4.38(s,6H),1.44(s,26H);MS(ESI):392(M+H).pound20.Pale yellow solid;Yield:81.5%;1H NMR (300MHz,CDCl3)d7.63(dd,J=7.4, 1.8Hz,1H),7.41(d, J=7.8Hz,1H),7.25–7.11(m,2H),6.81(dd,J=7.2,2.1Hz,1H), 6.31(d,J=2.1Hz,1H),6.24(d,J=7.7Hz,1H),6.01(d,J=2.1Hz, 1H),5.24(s,2H),3.94(s,3H),3.71(s,3H);MS(ESI):374(M+H).4.5.2.Coupling products22–39The structures of known coupling products23,25,27,28,32, 33,35,36and39were elucidated by comparison the spectral data with those reported in the literatures[11,13,27,30,31,33].pound22.White solid;1H NMR(300MHz,CDCl3)d 7.70(d,J=7.6Hz,1H),7.63(dd,J=7.7, 1.5Hz,1H),7.38(td, J=7.6, 1.5Hz,1H),7.31–7.26(m,2H),7.19(d,J=7.4Hz,1H), 7.02(t,J=7.7Hz,1H), 5.07(s,2H), 1.43(s,9H);13C NMR (75MHz,CDCl3)d153.61,139.03,131.80,130.83,128.35,127.33, 126.71,124.37,123.96,122.53,121.68,121.65,67.60,34.82, 29.80(3);EI-MS m/z(rel.int.,%):238(37),223(100),195(22), 165(22);HRMS(ESI):calculated for C17H18O+H=239.1436, found:239.1480.pound24.Pale yellow solid;1H NMR(300MHz,CDCl3) d7.95(dd,J=7.8,1.5Hz,1H),7.81(dd,J=8.2,1.5Hz,1H),7.70(d, J=7.4Hz,1H),7.46–7.35(m,2H),7.24–7.18(m,1H),7.13(t, J=8.0Hz,1H),5.26(s,2H);13C NMR(75MHz,CDCl3)d154.67, 146.11,136.73,135.13,135.08,134.29(s),133.09,133.84, 131.90,131.00,128.58,127.35,83.61,83.19,82.76,75.13;MS (ESI):228.05(M+H);HRMS(ESI):calculated for C13H9NO3+H=228.0661,found:228.0648.pound26.White solid;1H NMR(300MHz,CDCl3)d 7.95(d,J=2.2Hz,1H),7.79(d,J=7.6Hz,1H),7.68–7.57(m,2H), 7.49–7.29(m,6H),7.19(d,J=7.0Hz,1H),7.08(d,J=8.4Hz,1H), 5.17(s,2H);13C NMR(75MHz,CDCl3)d154.30,140.92,135.30, 131.40,129.98,128.74,128.45,128.22,127.77,126.90,126.84, 124.67,123.04,121.99,117.66,77.41,76.99,76.56,68.52;EI-MS m/z(rel.int.,%):258(100),257(85),228(17),229(14),215(6); HRMS(ESI):calculated for C19H14O+H=259.1123,found: 259.1101.pound29.White solid;1H NMR(300MHz,CDCl3)d 9.95(s,1H),7.89(d,J=1.7Hz,1H),7.76(d,J=7.6Hz,1H),7.50–7.30(m,3H),7.19(d,J=7.6Hz,1H),5.30(s,2H),3.98(s,3H); 13C NMR(75MHz,CDCl3)d190.79,149.62,149.08,130.79, 130.42,130.16,128.65,128.48,124.61,123.21,122.22,119.96, 109.58,68.87,56.04;MS(ESI):241.05(M+H);HRMS(ESI):calcu-lated for C15H12O3+H=241.0865,found:241.0847.pound30.White solid;1H NMR(300MHz,CDCl3)d 7.89(dd,J=7.7,1.6Hz,1H),7.75(dd,J=7.8,1.6Hz,1H),7.68(d, J=7.4Hz,1H),7.39(td,J=7.5, 1.1Hz,1H),7.32(td,J=7.4, 1.2Hz,1H),7.18(d,J=7.3Hz,1H),7.08(t,J=7.8Hz,1H),5.20(s, 2H), 3.92(s,3H);13C NMR(75MHz,CDCl3)d166.27,154.48, 134.84,133.30,131.13,129.22,128.50,128.06,127.16,124.60, 124.52,124.12,122.13,121.20,68.56,52.03;MS(ESI):241.05 (M+H);HRMS(ESI):calculated for C15H12O3+H=241.0865, found:241.0848.pound31.White solid;1H NMR(300MHz,CDCl3)d 8.40(d,J=2.1Hz,1H),7.85(dd,J=8.5, 2.1Hz,1H),7.81(d, J=7.7Hz,1H),7.42(t,J=7.1Hz,1H),7.33(td,J=7.4,1.1Hz,1H), 7.17(d,J=7.4Hz,1H),7.02(d,J=8.5Hz,1H),5.20(s,2H),2.62 (s,3H);13C NMR(75MHz,CDCl3)d196.68,158.64,131.38, 130.59,130.07,128.90,128.61,128.22,124.58,123.74,122.44, 122.10,117.25,68.48,26.31;MS(ESI):225.05(M+H);HRMS (ESI):calculated for C15H12O2+H=225.0916,found:225.0894.pound34.Pale solid;1H NMR(300MHz,CDCl3)d7.77 (d,J=7.6Hz,1H),7.61(d,J=7.8Hz,1H),7.46(d,J=7.5Hz,1H), 7.41(t,J=7.6Hz,1H),7.30(dd,J=11.3, 4.4Hz,2H),7.20(t, J=6.9Hz,1H),7.11(t,J=7.5Hz,1H),5.05(s,2H),2.57(s,3H);13C NMR(75MHz,CDCl3)d141.63,140.01,133.59,133.55,L.Zhang et al./Ultrasonics Sonochemistry19(2012)250–256255133.04,127.92,126.23,123.45,121.36,120.75,119.49,118.72, 108.98,101.90,47.94,8.94;MS(ESI):220.05(M+H);HRMS (ESI):calculated for C16H13N+H=220.1126,found:220.1104.pound37.White solid;1H NMR(300MHz,CDCl3)d 7.73–7.63(m,2H),7.38(t,J=7.6Hz,1H),7.30(dd,J=7.4,1.2Hz, 1H),7.24(d,J=9.6Hz,2H),7.16(d,J=6.8Hz,1H),7.02(t, J=7.6Hz,1H),5.14(s,2H),4.34(s,2H),1.44(s,9H);13C NMR (75MHz,CDCl3)d155.82,152.59,131.01,129.90,129.26,128.45, 127.64,127.32,124.53,122.74,122.55,122.14,121.75,79.17, 68.37,40.08,28.36;MS(ESI):312.15(M+H);HRMS(ESI):calcu-lated for C19H21NO3+H=312.1600,found:312.1571.pound38.Pale solid;1H NMR(300MHz,CDCl3)d7.68 (d,J=7.3Hz,1H),7.62–7.42(m,3H),6.56(s,1H),6.23(s,2H),5.12 (s,2H),3.91(s,6H);13C NMR(75MHz,CDCl3)d178.09,162.94, 161.79,149.55,142.33,137.66,133.34,130.71,128.42,122.79, 121.67,110.54,103.14,94.55,89.26,56.01,55.60,54.41;MS (ESI):294.05(M+H);HRMS(ESI):calculated for C18H15NO3+H=294.1130,found:294.1096.AcknowledgementThis work was supported by973Program(2007CB714504)and National Natural Science Foundation of China(20821063, 90913023).The authors thank Dr.Yin Ding(Nanjing University, China)for HRMS measurements.References[1]M.C.Kozlowski,B.J.Morgan,E.C.Linton,Total synthesis of chiral biaryl naturalproducts by asymmetric biaryl coupling,Chem.Soc.Rev.38(2009)3193–3207.[2]H.Guinaudeau,M.Leboeuf,A.Cave,Aporphine alkaloids.2,J.Nat.Prod.42(1979)325–360.[3]J.M.Brunel,BINOL:a versatile chiral reagent,Chem.Rev.105(2005)857–897.[4]R.Noyori,H.Takaya,BINAP–an efficient chiral element for asymmetriccatalysis,Acc.Chem.Res.23(1990)345–350.[5]J.Hassan,M.Sevignon, C.Gozzi, E.Schulz,M.Lemaire,Aryl–aryl bondformation one century after the discovery of the Ullmann reaction,Chem.Rev.102(2002)1359–1469.[6]T.Satoh,M.Miura,Catalytic direct arylation of heteroaromatic compounds,Chem.Lett.36(2007)200–205.[7]D.Alberico,M.E.Scott,utens,Aryl–aryl bond formation by transition-metal-catalyzed direct arylation,Chem.Rev.107(2007)174–238.[8]G.B.Bajracharya,O.Daugulis,Direct transition-metal-free intramoleculararylation of phenols,Org.Lett.10(2008)4625–4628.[9]D.D.Hennings,S.Iwasa,V.H.Rawal,Anion-accelerated palladium-catalyzedintramolecular coupling of phenols with aryl halides,.Chem.62(1997) 2–3.[10]F.Bellina,R.Rossi,Recent advances in the synthesis of(hetero)aryl-substitutedheteroarenes via transition metal-catalysed direct(hetero)arylation of heteroarene C–H bonds with aryl halides or pseudohalides,diaryliodonium salts,and potassium aryltrifluoroborates,Tetrahedron65(2009)10269–10310.[11]L.C.Campeau,M.Parisien,M.Leblanc,K.Fagnou,Biaryl synthesis via directarylation:establishment of an efficient catalyst for intramolecular processes,J.Am.Chem.Soc.126(2004)9186–9187.[12]M.Parisien, D.Valette,K.Fagnou,Direct arylation reactions catalyzed byPd(OH)(2)/C:evidence for a soluble palladium catalyst,.Chem.70(2005) 7578–7584.[13]L.C.Campeau,M.Parisien,A.Jean,K.Fagnou,Catalytic direct arylation witharyl chlorides,bromides,and iodides:intramolecular studies leading to new intermolecular reactions,J.Am.Chem.Soc.128(2006)581–590.[14]B.P.Mason,K.E.Price,J.L.Steinbacher,A.R.Bogdan,D.T.McQuade,Greenerapproaches to organic synthesis using microreactor technology,Chem.Rev.107(2007)2300–2318.[15]J.C.Brandt,S.C.Elmore,R.I.Robinson,T.Wirth,Safe and efficient ritterreactions inflow,Synlett(2010)3099–3103.[16]P.Watts,S.J.Haswell,The application of micro reactors for organic synthesis,Chem.Soc.Rev.34(2005)235–246.[17]S.L.Poe,M.A.Cummings,M.R.Haaf, D.T.McQuade,Solving the cloggingproblem:precipitate-forming reactions inflow,Angew.Chem.Int.Ed.45 (2006)1544–1548.[18]P.Cintas,J.L.Luche,Green chemistry–The sonochemical approach,GreenChem.1(1999)115–125.[19]R.Rajagopal,D.V.Jarikote,K.V.Srinivasan,Ultrasound promoted Suzuki cross-coupling reactions in ionic liquid at ambient conditions,mun.(2002)616–617.[20]J.T.Li,J.F.Han,J.H.Yang,T.S.Li,An efficient synthesis of3,4-dihydropyrimidin-2-ones catalyzed by NH2SO3H under ultrasound irradiation,Ultrason.Sonochem.10(2003)119–122.[21]J.Jin,M.M.Cai,J.X.Li,Highly efficient suzuki coupling of aryl chlorides in acontinuousflow capillary microreactor,Synlett(2009)2534–2538.[22]D.E.Ames, A.Opalko,Palladium-catalyzed cyclization of2-substitutedhalogenoarenes by dehydrohalogenation,Tetrahedron40(1984)1919–1925.[23]J.H.Li,W.J.Liu,Y.X.Xie,Recyclable and reusable Pd(OAc)(2)/DABCO/PEG-400system for Suzuki–Miyaura cross-coupling reaction,.Chem.70(2005) 5409–5412.[24]S.Koda,T.Kimura,T.Kondo,H.Mitome,A standard method to calibratesonochemical efficiency of an individual reaction system,Ultrason.Sonochem.10(2003)149–156.[25]T.Noël,J.R.Naber,R.L.Hartman,J.P.McMullen,K.F.Jensen,S.L.Buchwald,Palladium-catalyzed amination reactions inflow:overcoming the challenges of clogging via acoustic irradiation,Chem.Sci.2(2011)287–290.[26]G.B.Liu,J.L.Xu,C.C.He,G.Chen,Q.Xu,H.X.Xu,J.X.Li,Synthesis and evaluationof a novel series of quinoline derivatives with immunosuppressive activity, Bioorgan.Med.Chem.17(2009)5433–5441.[27]france,pointe,K.Fagnou,Mild and efficient palladium-catalyzedintramolecular direct arylation reactions,Tetrahedron64(2008)6015–6020.[28]J.Forrester,R.V.H.Jones,L.Newton,P.N.Preston,Synthesis and reactivity ofbenzylic sulfonium salts:benzylation of phenol and thiophenol under near-neutral conditions,Tetrahedron57(2001)2871–2884.[29]B.J.Margolis,J.J.Swidorski, B.N.Rogers,An efficient assembly ofheterobenzazepine ring systems utilizing an intramolecular palladium-catalyzed cycloamination,.Chem.68(2003)644–647.[30]Y.Antonio,M.E.Delacruz,E.Galeazzi,A.Guzman,B.L.Bray,R.Greenhouse,L.J.Kurz,D.A.Lustig,M.L.Maddox,J.M.Muchowski,Oxidative radical cyclization to pyrroles under reducing conditions–reductive desulfonylation of alpha-sulfonylpyrroles with tri-n-butyltin hydride,Can.J.Chem.72(1994)15–22.[31]N.Barbero,R.SanMartin,E.Dominguez,Divergent synthesis of isoindolo2,1-aindole and indolo1,2-a indole through copper-catalysed C-and N-arylations, Tetrahedron Lett.50(2009)2129–2131.[32]S.R.Flanagan,D.C.Harrowven,M.Bradley,Radical cyclisation reactions withindoles,Tetrahedron Lett.44(2003)1795–1798.[33]D.E.Ames, A.Opalko,Synthesis of dibenzofurans by palladium-catalyzedintramolecular dehydrobromination of2-bromophenyl phenyl ethers, Synthesis-Stuttgart(1983)234–235.256L.Zhang et al./Ultrasonics Sonochemistry19(2012)250–256。
2008 world journal of microbiology and biotechnology
ORIGINAL PAPERPurification and characterization of a thermostable uricase from Microbacterium sp.strain ZZJ4-1Lei Kai ÆXiao-Hang Ma ÆXue-Lai Zhou ÆXiao-Ming Jia ÆXia Li ÆKang-Ping GuoReceived:10April 2007/Accepted:30June 2007/Published online:25July 2007ÓSpringer Science+Business Media B.V.2007Abstract In order to study the properties of a thermo-stable uricase produced by Microbacterium sp.strain ZZJ4-1,the enzyme was purified by ammonium sulfate precipitation and DEAE-cellulose ion exchange,hydro-phobic and molecular sieve chromatography.The molec-ular mass of the purified enzyme was estimated to be 34kDa by SDS-PAGE.The enzyme was stable between pH 7.0and 10.00.The optimal reaction temperature of the enzyme was 30°C at pH 8.5.The K m and K cat of the enzyme were 0.31mM and 3.01s –1,respectively.Fe 3+could enhance the enzyme activity,whereas Ag +,Hg 2+,o -phenanthroline and SDS inhibited the activity of the enzyme considerably.After purification,the enzyme was purified 19.7-fold with 31%yield.As compared with uri-cases from other microbial sources,the purified enzyme showed excellent thermostability and other unique char-acteristics.The results of this work showed that strains of Microbacterium could be candidates for the production of a thermostable uricase,which has the potential clinical application in measurement of uric acid.Keywords Uricase ÁThermostability ÁMicrobacterium .ÁPurification ÁCharacterization ÁUric acidIntroductionUricases (urate oxidoreductase;EC 1.7.3.3),which catalyse the oxidative breakdown of uric acid,belong to a group ofenzymes in the purine degradation pathway found in ani-mals (Keilin 1959;Wallrath and Friedman 1991),plants (Montalbini et al.1997),fungi (Montalbini et al.1999),yeasts (Adamek et al.1990;Hongoh et al.2000;Koyama et al.1996)and bacteria (Yamamoto et al.1996).Since Bongaert et al.(Bongaert et al.1978)found that Bacillus fastidiosus could produce uricase and use uric acid as the only carbon source for growth in 1978,many reports on bacterial and yeast uricases,such as those from Arthrob-acter globiformis (Nobutoshi et al.2000),Bacillus subtilis (Hunag and Wu 2004),Candida utilis (Liu et al.1994)and Pseudomonas aeruginosa (Ishikawa et al.2004),have been published.In the human body,uric acid is a final product of purine catabolism and is excreted out of the body by the kidney.When the level of uric acid in blood increases over the normal value,it can lead to a group of diseases,such as gout (Nakagawa et al.2006),idiopathic calcium urate nephrolithiasis (Masseoud et al.2005)and renal failure (Capasso et al.2005),and it was also reported that a high level of uric acid was related to leukemia in children (Larsen &Loghman-Adham 1996).Consequently,uric acid concentration is an important parameter monitored in urine and blood samples in routine clinical examinations.At present,the colorimetric method that employs uricase and peroxidase is widely accepted as a simple,sensitive and highly specific test for uric acid examination.In this enzymatic system of uric acid analysis,uricase plays an important role:uric acid À!uricase5Àhydroxyisourate þH 2O 2þCO 2By determining the amount of peroxide formed in the reaction,the concentration of uric acid can be estimated.For the best result in this examination,the properties of theL.Kai ÁX.-H.Ma (&)ÁX.-L.Zhou ÁX.-M.Jia ÁX.Li ÁK.-P.GuoCollege of Life Sciences,Zhejiang University,Yuhangtang Road 388#,Hangzhou,Chinae-mail:maxiaohong@World J Microbiol Biotechnol (2008)24:401–406DOI 10.1007/s11274-007-9489-1uricase in this reaction system are important,especially its stability,which will determine the precision of the mea-surement.At present,most enzymes applied in the clinical test are used in solution,and most proteins,including this enzyme,are relatively unstable when dissolved in aqueous solution.Therefore,for the enzymatic examination appli-cation,research was undertaken to search for a thermostable enzyme(Guo et al.2006;Huang et al.1998;Zhou et al.2005).As mentioned above,there were many uricases that have been isolated from microorganisms,but the thermostability of the published uricases were relatively low(Suzuki et al. 2004).The most thermostable uricase was reported by Suzuki,but it would lose its activity after a short period of treatment at60°C(Suzuki et al.2004).This low stability is a disadvantage in clinical applications.In a previous study,we isolated a bacterium Micro-bacterium sp.strain ZZJ4-1that produced a thermostable uricase.The enzyme was stable at65°C and its solution retained its original activity even after storage at37°C for 40days(Zhou et al.2005).Considering that this enzyme has a potential value in practical application,the present study was undertaken to purify and study the properties of this new enzyme.Materials and methodsMaterials and chemicalsThe culture of Microbaterium sp.(strain ZZJ4-1)was maintained in our laboratory(Zhou et al.2005).The protein standards for SDS-PAGE were purchased from Invitrogen(Shanghai,China).All other chemicals used were of reagent or molecular biology grade and pur-chased from Hangzhou Huadong Medicine Group Co.,Ltd (Hangzhou,China).Cultivation conditionsThe fermentation medium consisted of3.0g of uric acid, 10.0g of maize milk,0.5g of MgSO4Á7H2O,0.5g of KH2PO4,2.0g of K2HPO4Á3H2O,0.1g of NaCl,1.0l of tap water and the pH was adjusted to7.5(Zhou et al. 2005).To cultivate the strain for production of the uricase, a loop of bacteria from a slant was inoculated into a500ml flask containing100ml liquid medium and incubated at 30°C for30h with a rotary shaker at120rev/min.Enzyme assay and protein measurementThe principle of enzyme measurement was as follows: uricase can catalyse the oxidation of uric acid to form 5-hydroxyisourate and H2O2,which is then measured using a reaction system containing4-aminoantipyrine,phenol and peroxidase as chromogens.In practical analysis, 0.10ml enzyme solution was incubated with a mixture of 0.6ml0.1M sodium borate buffer(pH8.5)containing 2mM uric acid,0.15ml of30mM4-aminoantipyrine, 0.1ml of1.5%phenol and0.05ml peroxidase(15U/ml) at37°C for20min(Masaru1981).The reaction was stopped by addition of1.0ml ethanol and the absorbance at540nm was read against the blank in a spectropho-tometer.One unit of enzyme was defined as the amount of enzyme that produces1.0l mole of H2O2per minute under the standard assay conditions.The protein was measured by the Folin-phenol method (Lowry et al.1951).Enzyme purificationUnless otherwise stated,all of the enzyme purification processes were performed with0.1M phosphate buffer at pH7.0(buffer A)at4°C.Cells from10.0l of media were harvested by centrifu-gation and washed twice with50mM phosphate buffer(pH 7.5),re-suspended in buffer A and disrupted by ultrasonic oscillation(120W oscillating for3s with6s intervals, repeated100times).After the cell debris had been sepa-rated by centrifugation,solid ammonium sulfate was added to the enzyme solution and the precipitate of the fractions from55%to80%saturation was collected by centrifuga-tion(8000rpm,40min,4°C).The enzyme was then dis-solved in a small amount of buffer(2:1,volume of the buffer/weight of the precipitate),dialyzed against0.01M phosphate buffer until the ammonium sulfate was removed.The dialyzed enzyme solution was put onto a DEAE-Cellulose column(5.5·50cm)previously equilibrated with buffer A and it was then eluted with2.0l of buffer A with a linear gradient of0–1.5M KCl(flow rate:2ml/ min).The fractions containing enzyme activity were col-lected.After the ammonium sulfate had been added to65% saturation,the enzyme solution was loaded onto a Toyo-pearl HW-65column(5.5·85cm)equilibrated previ-ously with buffer A containing65%ammonium sulfate. The enzyme was eluted with2.0l of the same buffer with a linear gradient of ammonium sulfate from65%to0%(flow rate:2ml/min).When the fractions containing enzyme activity had been collected,the ammonium sulfate was added to get85%saturation.The precipitate of the enzyme was then collected by centrifugation,dissolved in a small amount of buffer A,dialysed with the same buffer con-taining0.1M KCl and applied to a Sephadex G-75column (5.5·100cm)equilibrated with the same buffer.The enzyme was then eluted with2.0l of the same buffer(flow rate:2ml/min)and the fractions containing the highest specific activity were collected for further study.Characterization of enzymeTo study the effect of pH on the activity of uricase,the enzyme was assayed at different pHs in the range from4.0 to11.0with intervals of0.5.The following buffers were used:100mM citrate for pH4.0–6.0,100mM phosphate for pH6.0–8.5and100mM borate for pH8.5–11.The pH stability was studied by incubating the purified enzyme solution in the corresponding buffers in the range from4.0 to11.0at25°C for18h and measuring the residual activity.To study the effect of temperature on the uricase activity,the standard enzyme reaction solution was pre-incubated at temperatures of20–60°C with5°C intervals for5min and the enzyme solution was then added and incubated for20min at the same temperature to measure its activity.For thermostability testing,the purified uricase solution was incubated at temperatures in the range from 20°C to80°C for30min and the remaining activity was then measured.The apparent K m of the uricase was estimated by the double reciprocal plot method.At different concentrations of uric acid,the enzyme activity was assayed and the Km was calculated by the Lineweaver-Burk plotting according to the Michaelis-Menten equation.To study the effects of chemicals on the uricase activity, the enzyme solution was pre-incubated with the chemicals for30min at room temperature in phosphate buffer and the remaining uricase activity was assayed with the standard reaction system containing the corresponding chemical.The relative molecular mass of the enzyme was deter-mined by SDS-PAGE with10%polyacrylamide gels (Laemmli1970).The purity was determined by the specific activity of the enzyme and PAGE with10%polyacryl-amide gels.The protein was stained with0.1%Coomassie brilliant blue R250in4:1:5methanol/acetic acid/water (vol/vol/vol)solution and destained in the same solution without dye.ResultsPurification of the enzymeIn thefirst step of the purification,ammonium sulfate precipitation was applied.The amount of protein and the enzyme activity of each fraction were measured.The fractions with ammonium sulfate concentrations from65% to80%had the highest enzyme specific activity(0.43U/ mg),while fractions from35–55,55–65to80–85had the specific activity of0.02U/mg,0.09U/mg and0.07U/mg, respectively.The fractions with concentrations from65% to80%were collected,dialyzed and loaded onto a DEAE-cellulose column.The enzyme was eluted with the same buffer containing a linear concentration gradient of KCl from0M to1.5M.Enzyme activity was found in fractions from75to130and fractions from80to105were pooled, solid ammonium sulfate was added to65%saturation and the solution was loaded onto a Toyopearl HW65-C col-umn.The enzyme was then eluted with the same buffer with a decreasing linear concentration of ammonium sul-fate from65%to0%.The enzyme activity was found in the fractions from100to160and fractions from110to135 were pooled,concentrated and applied to the Sephadex G-75column.After being washed with buffer A containing 0.1M NaCl,the enzyme activity was observed in the fractions from30to80.The fractions from50to60were pooled as purified enzyme for further study.The purity of the enzyme after each purification step was examined by SDS-PAGE and is shown in Figure1. The results of the purification process are summarized in Table1.During the purification,the enzyme was purified 19.7-fold with a recovery of31%and the purified enzyme had a specific activity of5.32U mg-1.Molecular mass determinationThe purified enzyme showed a single protein band in SDS-PAGE and its molecular mass was estimated to be34kDa (Fig.2).Optimum reaction temperature and thermostability ofthe purified enzymeThe purified enzyme was stable at a relative high temper-ature.As shown in Fig.3,it was stable at65°C and it retained64%of its original activity even after beingtreated Fig.1SDS-PAGE pattern of uricase samples from different steps of purification.(A)crude extract;(B)ammonium sulfate precipitation;(C)DEAE-cellulose chromatography;(D)Toyopearl HW-65chro-matography;(E)Sephadex G-75chromatographyat70°C for30min.By comparison between the optimal reaction and stable temperature,it was shown that although the enzyme was stable at65°C,its optimum temperature was30°C,which was relatively low,and it only showed 21%relative activity at60°C.Optimum pH and the stability of the enzymeat different pHsThe activity of the enzyme was measured in different buffers with a pH range from4.0to11.0.The results showed that the enzyme had low activity at pH below5.5 or over10.5and had relatively high activity in the range from7.0to10.0,with the optimal reaction pH at8.5 (Fig.4).As shown in Fig.4,after being incubated in dif-ferent buffers at25°C for18h,the uricase was stable in the pH range from5.5to9.5.Kinetics and effect of chemicals on the activityof the enzymeThe K m of the purified enzyme for uric acid was estimated to be0.31mM,which was calculated from the slopes and intercepts of the regression lines of the Lineweaver-Burk plot by determining the enzyme activity at37°C and the K cat of the enzyme was estimated to be3.01s–1.The effects of different chemicals on the activity of the enzyme are summarized in Table2.It was shown that among the metal ions,Li+,Ag+and Hg+greatly inhibitedTable1Summary of the uricase purification process Purification steps Activity(U)Total protein(mg)Specific activity(U mg–1)Purification(fold)Yeild(%)Crude enzyme6012220400.271100 Ammoniumsulfate488595190.51 1.9081 DEAE-cellulose338540840.83 3.0756 Toyopearl HW-6528451530 1.86 6.8947 Sephdax G-751866351 5.3219.7031 Fig.2SDS-PAGE electrophoregram of uricase.M:Standard ProteinMarker;U:Uricasethe enzyme activity.The strongest suppression was ob-served in the case of Hg+,which suppressed almost all of the activity of this enzyme.The chelating reagents had different effects;EDTA had no inhibitory effect,whereas o-phenanthroline(OPT)could inhibit the activity of uricase considerably.DiscussionAt present,the uricases from many microorganisms have been studied and some gene sequences of uricases have been cloned and studied.It was shown that uricases belong to a group of enzymes that have the same catalytic char-acter,but a great diversity of molecular structures.Uricases from different sources may have different molecular mas-ses and amino acid sequences.In this study,the molecular mass of the uricase from strain ZZJ4-1was estimated to be 34kDa by SDS-PAGE,whereas uricases produced by Candida utilis(Koyama et al.1996)and Pseudomonas aeruginosa(Ishikawa et al.2004)had the molecular mas-ses of34and54kDa,respectively.The apparent K m value of this uricase was0.31mM,while uricases from Ar-throbacter,Bacillus sp.and Candida sp.had K m values of 75,75,46l M,respectively(Suzuki et al.2004).Some relatively thermostable uricases,such as those produced by Arthrobacter globiformis FERM BP-360, Bacillus sp.TB-90and Candida utili s,have been studied and these were stable at55or60°C(Suzuki et al.2004). In this work,the uricase produced by strain ZZJ4-1showed good thermostability.After incubation at65°C for30min, the enzyme from strain ZZJ4-1still retained98.9%of its original activity,and even after incubation at70°C for 30min,the remaining activity was64%.Additionally,the pH stability of the enzyme from strain ZZJ4-1was a little broader than that of the uricases from the above three species(Suzuki et al.2004).The effects of metal ions on the enzyme activity from strain ZZJ4-1were also compared with the above three enzymes(Suzuki et al.2004).Considering that some uri-cases require metal ion cofactors for activity(Wu et al. 1989;Chu et al.1996)and whether or not an enzyme is inhibited by certain metals is an important characteristic, experiments were carried out to examine the effect of metals on the activity of this enzyme.It was shown that the enzyme from strain ZZJ4-1was not inhibited by Cu2+,Fe3+ or Zn2+,while uricases from the other three strains were strongly inhibited by these ions.Ag+is a strong inhibitor of the uricases from strain ZZJ4-1,Candida utilis and Bacillus sp.TB-90,while it had no such effect on the uricase from Arthrobacter globiformis FERM BP-360. When EDTA,a chelating reagent,was added to the enzyme solution at afinal concentration of20mM,the activity of the uricase was only slightly inhibited.When the stronger metal-ion-chelating reagent o-phenanthroline was added to the enzyme solution,the activity of the enzyme was strongly inhibited.This phenomenon indicates that some yet unidentified metal ion is strongly bound in the enzyme and forms part of the uricase structure,which is very important to keep its catalytic activity.This property is also different from the uricase of Arthrobacter globiformis FERM BP-360(Suzuki et al.2004).It was shown that although the uricase from the strain ZZJ4-1was stable at65°C,the enzyme was at its optimal activity at30°C and it only showed21%of its maximal activity at60°C.This implied that the molecular structure of enzyme had a reversible change at a temperature be-tween30°and60°C,which had a negative effect on its catalysis activity.But considering that most clinical enzy-matic examinations are undertaken at25to37°C,this property will not affect its application in clinical exami-nation.ReferencesAdamek V,Suchova M,Demnerova K et al(1990)Fermentation of Candida utilis for uricase production.J Indu Microbiol6:85–90 Bongaerts GP,Uitzetter J,Brouns R et al(1978)Uricase of Bacillus fastidiosus.Properties and regulation of synthesis.Biochim Biophys Acta527:348–358Capasso G,Jaeger Ph,Robertson WG et al(2005)Uric acid and the kidney:urate transport,S tone disease and progressive renal failure.Curr Pharm Des11:4153–4159Table2Effects of chemicals on uricase activityChemicals Final Concentration Residual activity(%)Blank–100Fe3+1mM118.5Ca2+1mM107.7Ba2+1mM103.1Mn2+1mM101.5Zn2+1mM100.7Cu2+1mM98.9Li+1mM81.5Ag+1mM7.5Hg2+1mM 1.8NaN320mM101.2EDTA20mM99.8o-Phenanthroline2mM 1.2Tween200.10%(w/v)99.7Tween800.10%(w/v)80.5Triton X-1000.10%(w/v)99.7SDS0.50%(w/v)51.3Chu R,Lin Y,Usuda N,Rao MS et al(1996)Mutational analysis of the putative copperbinding site of rat urate oxidase.Ann NY Acad Sci804:777–780Guo K,Ma X,Sun G et al(2006)Expression and characterization of a thermostable sarcosine oxidase(SOX)from Bacillus sp.in Escherichia coli.Appl Microbiol Biotechnol73:559–566 Hongoh Y,Sasaki T,Ishikawa H(2000)Cloning,sequence analysis and expression in Escherichia coli of the gene encoding a uricase from the yeast-like symbiont of the brown planthopper, Nilaparvata lugens.Insect Biochem Mol Biol30:173–182 Huang HS,Kabashima T,Ito K et al(1998)Thermostable glycerol kinase from Thermusflavus:cloning,sequencing,and expression of the enzyme gene.Biochim Biophys Acta1382:186–190 Hunag S,Wu T(2004)Modified colorimetric assay for uricase activity and a screen for mutant Bacillus subtilis uricase genes following StEP mutagenesis.Eur J Biochem271:517–523 Ishikawa J,Yamashita A,Mikami Y et al(2004)The complete genomic sequence of Nocardia farcinica IFM10152.Proc Natl Acad Sci USA101:14925–14930Keilin J(1959)The biological significance of uric acid and guanine excretion.Biol Rev34:265–296Koyama Y,Ichikawa T,Nakano E(1996)Cloning,sequence analysis, and expression in Escherichia coli of the gene encoding the Candida utilis urate oxidase(uricase).J Biochem(Tokyo), 120:969–973Laemmli UK(1970)Cleavage of structural proteins during the assembly of the head of bacteriophage T4.Nature227:680–685 Larsen G,Loghman-Adham M(1996)Acute renal failure with hyperuricemia as initial presentation of leukemia in children.J Pediatr Hematol Oncol18:191–194Liu J,Li G,Liu H et al(1994)Purification and properties of uricase from Candida sp.and its application in uric acid analysis in serum.Appl Biochem Biotechnol47:57–63Lowry OH,Rosebrough NJ,Farr AL et al(1951)Protein measure-ment with the Folin Phenol reagent.J Biol Chem193:265–275Masaru P(1981)Purification and some properties of Sarcosine Oxidase from Corynebacterium sp.U-96.J Biochem89:599–607 Masseoud D,Rott K,Liu-Bryan R et al(2005)Overview of hyperuricaemia and gout.Curr Pharm Des11:4117–4124 Montalbini P,Aguilar M,Ineda M(1999)Isolation and character-ization of uricase from bean leaves and its comparison with uredospore enzyme.Plant Sci147:139–147Montalbini P,Redondo J,Caballero JL(1997)Uricase from leaves: its purification and characterization from three different higher plants.Planta202:277–283Nakagawa T,Mazzali M,Kang DH et al(2006)Uric acid-a uremic toxin?Blood Purif24:67–70Nobutoshi K,Keisuke S,Takao M et al(2000)Determination of uric acid in plasma by closed-loopfia with a coimmobilized enzyme flow cell.Anal Sci16:1203–1205Suzuki K,Sakasegawa SI,Misaki H et al(2004)Molecular cloning and expression of uricase gene from Arthrobacter globiformis in Escherichia coli and characterization of the gene product.J Biosci Bioeng98:153–158Wallrath LL,Friedman TB(1991)Species differences in the temporal pattern of Drosophila urate oxidase gene expression are attrib-uted to trans-acting regulatory changes.Proc Natl Acad Sci USA 88:5489–5493Wu XW,Lee CC,Muzny DM et al(1989)Urate oxidase:primary structure and evolutionary implications.Proc Natl Acad Sci USA 86:9412–9416Yamamoto K,Kojima Y,Kikuchi T et al(1996)Nucleotide sequence of the uricase gene from Bacillus sp.TB-90.J Biochem119:80–84Zhou XL,Ma XH,Sun GQ(2005)Isolation of a thermostable uricase-producing bacterium and study on its enzyme production conditions.Process Biochem40:3749–3753。
SnCl2还原硝基 97%
2-Arylbenzoxazoles as CETP inhibitors:Substitution of the benzoxazole moietyCameron J.Smith a,*,Amjad Ali a ,Liya Chen a ,Milton L.Hammond a ,Matt S.Anderson b ,Ying Chen b ,Suzanne S.Eveland b ,Qiu Guo b ,Sheryl A.Hyland b ,Denise ot b ,Carl P.Sparrow b ,Samuel D.Wright b ,Peter J.Sinclair aa Department of Medicinal Chemistry,Merck Research Laboratories,Rahway NJ 07065,United States bDepartment of Cardiovascular Diseases,Merck Research Laboratories,Rahway NJ 07065,United Statesa r t i c l e i n f o Article history:Received 26June 2009Revised 23October 2009Accepted 26October 2009Available online 29October 2009Keywords:Cholesteryl ester transfer protein CETPHigh density lipoprotein HDLBenzoxazolea b s t r a c tA series of 2-arylbenzoxazole inhibitors of the cholesterol ester transfer protein (CETP)is described.Structure–activity studies focused on variation of the substitution of the benzoxazole moiety.Substitu-tion at the 5-and 7-positions of the benzoxazole moiety was found to be beneficial for CETP pound 47was found to be the most potent inhibitor in this series and inhibited CETP with an IC 50of 28nM.Ó2009Elsevier Ltd.All rights reserved.Coronary heart disease (CHD)is now the leading cause of death of people in developed countries.Elevated levels of low-density lipoprotein-cholesterol (LDL-C)has been identified as a major risk factor for CHD.The development of the statins has significantly helped to reduce LDL-C levels in patients at risk for CHD.1,2There is now a growing body of epidemiological evidence linking in-creased levels of high density lipoprotein-cholesterol (HDL-C)with decreased risk of development of CHD.3–6Some cholesterol lower-ing drugs,including niacin,fibrates and statins,have a modest ef-fect on increasing HDL-C levels.7–10Regardless,niacin remains the front line therapy for raising HDL-C levels despite its modest effi-cacy ($20%increase).Consequently there is a need for better ther-apies to address this problem.The beneficial effects of high density lipoprotein (HDL)are thought to arise from its participation in reverse cholesterol trans-port (RCT)as well as its anti-inflammatory and anti-oxidant prop-erties.11,12Cholesteryl ester transfer protein (CETP)mediates the exchange of cholesteryl ester (CE)from HDL with triglycerides pri-marily from very low-density lipoprotein (VLDL).11,12Inhibition of CETP would therefore be expected to increase serum HDL-C levels.Clinical studies in humans with the CETP inhibitors,dalcetrapib (JTT-705),13,14torcetrapib,15–19and anacetrapib 20–22established that pharmacological inhibition of CETP leads to significant in-creases in HDL-C concentrations.Despite this observation,imagingstudies with torcetrapib showed the compound had no effect on the progression of atherosclerosis.23–25Additionally,the torcetra-pib phase III trial was prematurely halted after the observation ofNO O F 3CF 3CCF 3O OF 3C33OFSNHOOdalcetrapib (JTT-705)torcetrapibanacetrapib0960-894X/$-see front matter Ó2009Elsevier Ltd.All rights reserved.doi:10.1016/j.bmcl.2009.10.099*Corresponding author.Tel.:+17325942529;fax:+17325949545.E-mail address:cameron_smith@ (C.J.Smith).Bioorganic &Medicinal Chemistry Letters 20(2010)346–349Contents lists available at ScienceDirectBioorganic &Medicinal Chemistry Lettersj o ur na l h om e pa ge :w w w.e ls e v ie r.c om /lo c at e/bm c lincreased mortality in patients receiving torcetrapib and atorva-statin relative to the atorvastatin-only group.19This adverse effect may have been related to the observation of an increase in mean systolic blood pressure in the torcetrapib treated arm.There is cur-rently significant debate over whether the adverse effects observed with torcetrapib were caused by mechanism based factors or off-target activities.26–28Despite the uncertainty regarding the viabil-ity of CETP inhibitors,there is continued interest in the develop-ment of a cardiovascular drug against this target.This communication details the identification and optimization of a ser-ies of2-arylbenzoxazole based CETP inhibitors.Inhibition of CETP mediated CE transfer was characterized in vitro using afluorescence transfer assay.29The assay uses syn-thetic HDL donor particles that contain self-quenching BODIPY la-beled CE along with an additionalfluorescence quencher.As the BODIPY labeled CE is transferred from the donor particle to an acceptor lipoprotein by CETP,fluorescence is observed and quanti-fied.Inhibition of CETP mediated CE transfer was characterized by a decrease in levels offluorescence observed relative to control.A high throughput screen of compounds in the Merck collection at2l M using the above assay was conducted and hits were con-firmed by titration.2-Arylbenzoxazole1was identified as a screen-ing hit.This lead class was also independently identified by researchers at Bristol–Myers Squibb and published subsequent to our studies.30Their work showed the importance of substitution at the benzoxazole5-position on potency of CETP inhibition.The work described in this publication is consistent with that described by BMS,but also shows that further substitution of the benzoxaz-ole moiety at the7-position leads to compounds with additional enhancement of CETP inhibition.O NH OON1ClThe development of lead compound1began with the investiga-tion of the effect of variation of the substitution of the benzoxazole moiety.The synthetic approach used is shown in Scheme1.Key carboxylic acid2was synthesized from phenoxyacetic acid3by first treating with oxalyl chloride to form the corresponding acid chloride4.Coupling with4-aminobenzoic acid afforded carboxylic acid2.Benzoxazoles were then synthesized by one of three meth-ods.31Activation of2as the corresponding acid chloride using oxa-lyl chloride followed by coupling with a range of substituted2-aminophenols afforded amides of the general structure6.A solu-tion of amides6in xylene was heated at reflux in a Dean–Stark apparatus with either p-tolenesulfonic acid or pyridinium p-tolu-enesulfonic acid,to afford2-arylbenzoxazoles of general structure 7.Alternatively,a solution of carboxylic acid2,a2-aminophenol and boric acid in xylene could be heated at reflux or subjected to microwave irradiation at270°C to afford the corresponding2-aryl-benzoxazole.2-Aminophenols were either commercially available or obtained by reduction of the corresponding2-nitrophenol using either heterogeneous palladium or platinum oxide catalyzed hydrogenation or treatment with tin(II)chloride(Scheme2).Some 2-nitrophenols were obtained by nitration of the corresponding phenols.The CETP inhibition data for a series of compounds of different benzoxazole phenyl ring substitution is shown in Table1and it can be seen that substitution of this ring appreciably alters CETP inhib-itory activity.The unsubstituted benzoxazole(8)was10-fold less active than lead compound1.A survey of a series of benzoxazole substituents(compounds9–22)showed a clear preference for sub-stitution at the5-position.In particular the5-nitro and5-cyanoTable1SAR of2-arylbenzoxazolesData reported is derived from duplicate wells and three independent experi-ments.Mean IC50values were determined from10-point,one-third log concen-tration response curves and standard errors were610%.b IC50not determined if%max inhibition was<50%.C.J.Smith et al./Bioorg.Med.Chem.Lett.20(2010)346–349347derivatives 15and 20were found to be the most potent CETP inhibitors with IC 50s of 0.94and 0.13l M,pounds 23–29(Table 2)represent a series of substitutions that are toler-ated at the 5-position with only methoxy (compound 24)showing better potency than the original lead 1.Other 5-substituents such as larger alkyl,trifluoromethyl,methyl ester,carboxylic acid,amides,carbamates,sulfonamides,sulfones,hydroxyl,anilino,amidine,tetrazole and substituted phenyl were found to have little or no inhibitory activity (data not shown).The best 5-substituted derivative found was therefore 5-cyano derivative 20and this rep-resented an early benchmark compound.This was consistent with the work published by researchers at Bristol–Myers Squibb.30Holding the cyano group constant,the SAR of additional benz-oxazole substitution was then explored (Table 3).The three possi-ble regioisomeric methyl derivatives 31,33and 35were prepared via palladium catalyzed cyanation of the corresponding aryl ha-lides 30,32and 34,respectively.The CETP inhibition data clearly shows that incorporation of a methyl group is preferred at the 7-position and affords a twofold increase in potency (compare 35to 20and 34to 23).Compound 35has a CETP IC 50of -pounds 36–41show that a number of other substituents are toler-ated at the 7-position in combination with either 5-cyano or 5-halo the most potent being 5-cyano-7-fluoro derivative 39with a CETP IC 50of 62nM.The substitution of the 7-position was further investigated by the synthesis of a series of alcohols (compounds 44–57,Table 4).These compounds were synthesized from acetophenone 40as shown in Scheme 3.Treatment of ketone 40with sodium borohy-dride or a Grignard reagent afforded secondary or tertiary alcohols respectively of general structure 42.These compounds were then transformed into nitriles of general structure 43via palladium cat-alyzed cyanation.By and large,potency of CETP inhibition is inver-sely proportional to the size of the alkyl group added to acetophenone 40,the best compound being methyl derivative 47with a CETP IC 50of 28nM.Replacement of the 5-cyano group of 47with a hydrogen to give compound 57results in a 10-fold loss in potency of CETP inhibition confirming the importance of substi-tution at both the 5-and 7-positions.Compounds 20,35and 47were evaluated in a pharmacody-namic model in mice expressing cynomolgus monkey CETP anddid not show an increase in HDL-C levels.This may be attributed to the lack of oral bioavailability observed with these compounds in mouse PK studies.In summary,after high throughput screening of the Merck com-pound collection identified 2-arylbenzoxazole 1as a lead,it was developed into compounds 35and 47using a modular synthetic approach that showed the importance of substitution at the 7-po-sition for potency enhancement over the 5-position alone.These compounds represent important leads for further development of this structure class as CETP inhibitors.Further modifications of the amide and aryloxy moieties will be reported.Table 2SAR of 5-substituted-2-arylbenzoxazolesData reported is derived from duplicate wells and three independent experi-ments.Mean IC 50values were determined from 10-point,one-third log concen-tration response curves and standard errors were 610%.bSynthesized by reduction of 26;NaBH 4,MeOH,room temperature,1h,quant.cSynthesized from 23by Stille coupling;vinyltributyl tin.(Ph 3P)4Pd,DMF,80°C,12h,13%.dSynthesized from 23by Sonagashira coupling;(i)TMS acetylene,Pd(PPh 3)2Cl 2,CuI,Ph 3P,Et 2NH,DMF,microwave irradiation,120°C,75min,(ii)aq NaOH,THF,room temperature,1h,32%.Table 3SAR of disubstituted-2-arylbenzoxazolesData reported is derived from duplicate wells and three independent experi-ments.Mean IC 50values were determined from 10-point,one-third log concen-tration response curves and standard errors were 610%.Table 4SAR of 5,7-disubstituted-2-arylbenzoxazolesData reported is derived from duplicate wells and three independent experi-ments.Mean IC 50values were determined from 10-point,one-third log concen-tration response curves and standard errors were 610%.bSynthesized by reduction of 40;NaBH 4,MeOH,room temperature,1h,quant.cSynthesized from 46;LiAlH 4,THF,room temperature,1.5h,14%.348 C.J.Smith et al./Bioorg.Med.Chem.Lett.20(2010)346–349References and notes1.Kearney,P.M.;Blackwell,L.;Collins,R.;Keech,A.;Simes,J.;Peto,R.;Armitage,J.;Baigent,ncet2008,371,117.2.Baigent,C.;Keech,A.;Kearney,P.M.;Blackwell,L.;Buck,G.;Pollicino,C.;Kirby,A.;Sourjina,T.;Peto,R.;Collins,R.;Simes,ncet2005,366,1267.3.Rhoads,G.;Gulbrandsen,C.;Kagan,A.N.Eng.J.Med.1976,294,293.4.Castelli,W.;Doyle,J.;Gordon,T.;Hames,C.;Hjortland,M.;Hulley,S.;Kagan,A.;Zukel,W.Circulation1977,55,767.5.Gordon,T.;Castelli,W.P.;Hjortland,M.C.;Kannel,W.B.;Dawber,T.R.Am.J.Med.1977,62,707.6.Lewington,S.;Whitlock,G.;Clarke,R.;Sherliker,P.;Emberson,J.;Halsey,J.;Qizilbash,N.;Peto,R.;Collins,ncet2008,370,1829.7.**JAMA1984,251,365.8.Brown,B.G.;Stukovsky,K.H.;Zhao,X.-Q.Curr.Opin.Lipidol.2006,17,631.9.Otvos,J. D.;Collins, D.;Freedman, D.S.;Shalaurova,I.;Schaefer, E.J.;McNamara,J.R.;Bloomfield,H.E.;Robins,S.J.Circulation2006,113,1556. 10.Pedersen,T.R.;Olsson,A.G.;Færgeman,O.;Kjekshus,J.;Wedel,H.;Berg,K.;Wilhelmsen,L.;Haghfelt,T.;Thorgeirsson,G.;Pyorala,K.;Miettinen,T.;Christophersen,B.;Tobert,J.A.;Musliner,T.A.;Cook,T.J.Circulation1998,97, 1453.11.Tall,A.R.;Yvan-Charvet,L.;Terasaka,N.;Pagler,T.;Wang,N.Cell Metab.2008,7,365.12.Barkowski,R.S.;Frishman,W.H.Cardiol.Rev.2008,16,154.13.de Grooth,G.J.;Kuivenhoven,J. A.;Stalenhoef, A. F.H.;de Graaf,J.;Zwinderman,A.H.;Posma,J.L.;van Tol,A.;Kastelein,J.J.P.Circulation2002, 105,2159.14.Kuivenhoven,J.A.;de Grooth,G.J.;Kawamura,H.;Klerkx,A.H.;Wilhelm,F.;Trip,M.D.;Kastelein,J.J.P.Am.J.Cardiol.2005,95,1085.15.Clark,R.W.Curr.Opin.Pharmacol.2006,6,162.16.Brousseau,M.E.;Schaefer,E.J.;Wolfe,M.L.;Bloedon,L.T.;Digenio,A.G.;Clark,R.W.;Mancuso,J.P.;Rader,D.J.N.Eng.J.Med.2004,350,1505.17.Davidson,M.H.;McKenney,J.M.;Shear,C.L.;Revkin,J.H.J.Am.Coll.Cardiol.2006,48,1774.18.McKenney,J.M.;Davidson,M.H.;Shear,C.L.;Revkin,J.H.J.Am.Coll.Cardiol.2006,48,1782.19.Barter,P.J.;Caulfield,M.;Eriksson,M.;Grundy,S.M.;Kastelein,J.J.P.;Komajda,M.;Lopez-Sendon,J.;Mosca,L.;Tardif,J.-C.;Waters,D.D.;Shear,C.L.;Revkin,J.H.;Buhr,K.A.;Fisher,M.R.;Tall,A.R.;Brewer,B.N.Eng.J.Med.2007,357,2109.20.Bloomfield,D.;Carlson,G.L.;Sapre,A.;Tribble,D.;McKenney,J.M.;Littlejohn,T.W.;Sisk,C.M.;Mitchel,Y.;Pasternak,R.C.Am.Heart J.2009,157,352. 21.Krishna,R.;Anderson,M.S.;Bergman,A.J.;Jin,B.;Fallon,M.;Cote,J.;Rosko,K.;Chavez-Eng, C.;Lutz,R.;Bloomfield, D.M.;Gutierrez,M.;Doherty,J.;Bieberdorf,F.;Chodakewitz,J.;Gottesdiener,K.M.;Wagner,ncet2007, 370,1907.22.Krishna,R.;Bergman,A.;Jin,B.;Fallon,M.;Cote,J.;Van,H.P.;Laethem,T.;Gendrano,I.I.;Van,D.K.;Hilliard,D.;Laterza,O.;Snyder,K.;Chavez-Eng,C.;Lutz,R.;Chen,J.;Bloomfield,D.;De,S.M.;Van,B.L.;Gutierrez,M.;Al-Huniti, N.;Dykstra,K.;Gottesdiener,K.;Wagner,J.Clin.Pharmacol.Ther.2008,84,679.23.Kastelein,J.J.P.;van,L.S.I.;Burgess,L.;Evans,G.W.;Kuivenhoven,J.A.;Barter,P.J.;Revkin,J.H.;Grobbee,D.E.;Riley,W.A.;Shear,C.L.;Duggan,W.T.;Bots,M.L.N.Eng.J.Med.2007,356,1620.24.Nissen,S.E.;Tardif,J.-C.;Nicholls,S.J.;Revkin,J.H.;Shear,C.L.;Duggan,W.T.;Ruzyllo,W.;Bachinsky,W.B.;Lasala,G.P.;Tuzcu,E.M.N.Eng.J.Med.2007, 356,1304.25.Bots,M.L.;Visseren,F.L.;Evans,G.W.;Riley,W.A.;Revkin,J.H.;Tegeler,C.H.;Shear,C.L.;Duggan,W.T.;Vicari,R.M.;Grobbee,D.E.;Kastelein,ncet 2007,370,153.26.Rader,D.J.N.Eng.J.Med.2007,357,2180.27.Tall,A.R.;Yvan-Charvet,L.;Wang,N.Arterioscler.Thromb.Vasc.Biol.2007,27,257.28.Tall,A.R.N.Eng.J.Med.2007,356,1364.29.Eveland,S.S.;Milot,D.P.;Guo,Q.;Chen,Y.;Hyland,S.A.;Peterson,L.B.;Jezequel-Sur,S.;O’Donnell,G.T.;Zuck,P.D.;Ferrer,M.;Strulovici,B.;Wagner, J.A.;Tanaka,W.K.;Hilliard,D.A.;Laterza,O.;Wright,S.D.;Sparrow,C.P.;Anderson,M.S.Anal.Biochem.2007,368,239.30.Harikrishnan,L.S.;Kamau,M.G.;Herpin,T.F.;Morton,G.C.;Liu,Y.;Cooper,C.B.;Salvati,M.E.;Qiao,J.X.;Wang,T.C.;Adam,L.P.;Taylor,D.S.;Chen,A.Y.A.;Yin,X.;Seethala,R.;Peterson,T.L.;Nirschl,D.S.;Miller,A.V.;Weigelt,C.A.;Appiah,K.K.;O’Connell,J.C.;Michael Lawrence,R.Bioorg.Med.Chem.Lett.2008,18,2640.31.Benzoxazole synthesis—General method1:A mixture of2(1.05mmol),2-aminophenol derivative(1.05mmol)and boric acid(1.37mmol)in o-xylene (60mL)was heated at reflux under a Dean–Stark apparatus overnight.After this time the reaction mixture was diluted with EtOAc(50mL),washed successively with saturated NaHCO3(50mL),H2O(50mL),and brine(50mL), dried(Na2SO4)and concentrated in vacuo to afford the crude product.This was purified byflash chromatography and/or reversed phase HPLC to afford the desired benzoxazole.General method2:A mixture of2(0.307mmol),2-aminophenol derivative(0.430mmol)and boric acid(0.430mmol)in o-xylene(2.5mL)was subjectedto microwave irradiation(300W,270°C,60min).The reaction mixture was diluted with EtOAc(25mL),washed successively with saturated NaHCO3 (25mL),H2O(25mL),and brine(25mL),dried(MgSO4)and concentrated in vacuo to afford the crude product.This was purified byflash chromatography and/or reversed phase HPLC to afford the desired benzoxazole.General method3:A solution of oxalyl chloride(2M in CH2Cl2,1.40mmol)was added to a stirred suspension of2(0.702mmol)in CH2Cl2(11mL)followed bya few drops of DMF at room temperature under N2.The reaction was stirred atroom temperature for4h after which time the suspension dissolved.The reaction mixture was concentrated in vacuo and azeotroped with toluene (10mL).The crude acid chloride and2-aminophenol(1.05mmol)were dissolved in1,4-dioxane(20mL)and heated at reflux for4h under N2.The reaction was diluted with EtOAc(50mL)and water(50mL)and the aqueous layer was extracted with EtOAc(2Â50mL).The combined organic extracts were washed with brine(50mL),dried(Na2SO4)and concentrated in vacuo to afford the crude amide product.A mixture of the crude amide and pyridinium p-toluenesulfonate(0.0702mmol)in o-xylene(30mL)was heated at reflux under a Dean–Stark apparatus overnight under N2.The reaction was diluted with EtOAc(100mL)and washed successively with saturated NaHCO3(50mL), water(50mL)and brine(50mL),dried(Na2SO4)and concentrated in vacuo to afford the crude product.This was purified byflash chromatography and/or reversed phase HPLC to afford the desired benzoxazole.C.J.Smith et al./Bioorg.Med.Chem.Lett.20(2010)346–349349。
核酸适配体
a b s t r a c t
Herein, we have successfully built up connections between nanoparticles and nanoclusters, and further constructed a surface-enhanced fluorescence (SEF) strategy based on the two types of nanomaterials for selectively assaying carcinoembryonic antigen (CEA). Specifically, silver nanoclusters provided the original fluorescence signal, while gold nanoparticles modified with DNA served as the fluorescence enhancer simultaneously. On the basis of this proposed nano-system, the two nanomaterials were linked by CEA–aptamer, thus facilitating SEF occurring. Nevertheless, more competitive interactions between CEA and CEA–aptamer emerged once CEA added, leading to SEF failed and their fluorescence decreased. Significantly, this creative method was further applied to detect CEA, and showed the linear relationship between the fluorescence intensity and CEA concentrations in the range of 0.01–1 ng mL À 1 with a detection limit of 3 pg mL À 1 at a signal-to-noise ratio of 3, demonstrating its sensitivity and promising towards multiple applications. On the whole, this approach we established may broaden potential ways of combining nanoparticles and nanoclusters for detecting trace targets in bioanalytical fields. & 2014 Elsevier B.V. All rights reserved.