BE试验一般程序

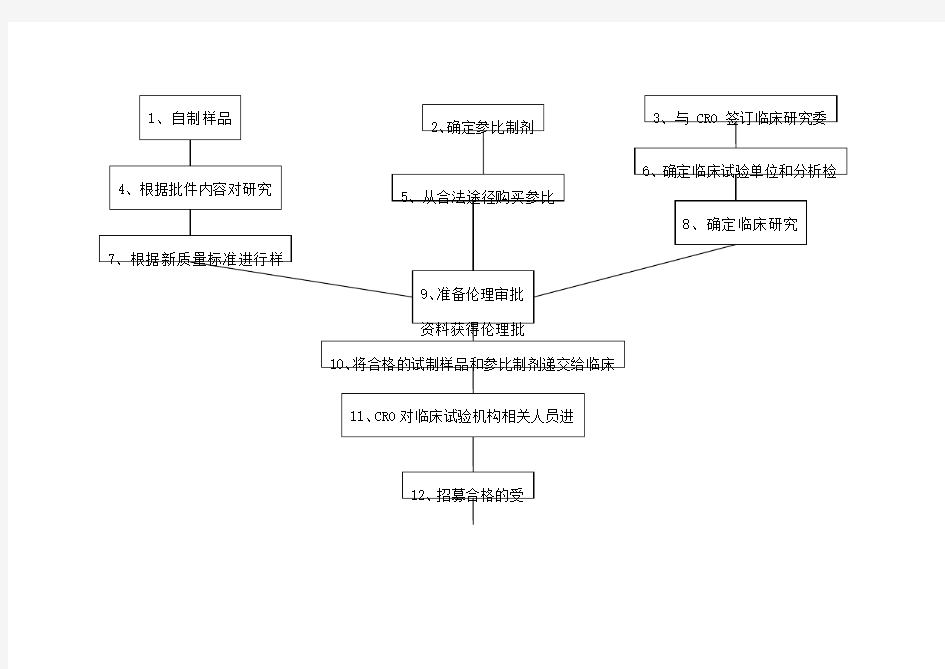
生物等效试验流程:
所做工作流程及所需信息资料:
1、与原研参比制剂进行多种介质溶出曲线比较:0.1M HCl(pH1.2)、
因子法测得pH4.5醋酸-醋酸钠缓冲液、pH6.8磷酸盐缓冲液、H2O中,f
2
受试样品和参比样品在15分钟的平均溶出量均不低于85%。
2、根据原研已在境内上市,所以确定已进口的原研样品作为参比制剂。
3、与CRO签订委托合同,该合同应包括双方的权利义务,明确分为自制样品本身所造成的不等效和非药物导致的不等效,自制样品本身所致不等效:药物溶解性、药物吸收性、晶型、化学稳定性、处方工艺等;非药物本身所致不等效(试验设计):试验设计样本量、测定方法学问题、生物样本采集点选择、统计分析方法错误;CRO费用分为预BE和正式试验,采用分期付款方式;是否在合同中加强CRO对临床机构的监督检查
4、基于批件内容,对涉及质量标准变更所需增加的研究内容,应尽快完成,以便得到最终确定的质量标准以进行BE样品试制;对不涉及质量标准变更的研究内容可以变进行研究变做BE,在完成BE最终将申报资料报到国家局之前完成。同时相关BE样品试制所采购的原辅料质量标准和检验均应升级到申报所对应的中国药典标准。
5、合法途径采购参比制剂,选定代理商所提供的资质:经营资质,供货合同或协议,发票,检验报告,销售清单。发票最好开给以药厂名义申报的药厂,检验报告最好能要到原研的检验报告(GSP的销售商肯定有,就看他们愿不愿意提供),如果拿不到经销商的检验报告,应由申报的药厂进行参比制剂的检验工作,并出具检验报告。
6、确定临床机构,根据项目特点和既往同类项目操作经验选择临床激光和分析检测机构。原则是专业、经验丰富,并且要考虑CRO与临床机构的关系是否紧密;如果CRO有分析测试部门,考察其专业性和规范性,如果CRO没有分析检测部分,应确定其合作的分析测试单位的专业性、规范性和经验。
7、BE试验样品试制,在GMP车间生产受试样品应来自一个不少于生产规模1/10的批次或100000单位,两者中选择更多,除非另外说明理由;使用的生产批次应该确实保证产品和过程在工业规模可行。试验药品的包装:应该对每位受试者和每个周期分别包装参比样品和受试药品,在它们被运往试验地点之前或在试验地点进行包装。包装(包括标签)应按照GMP规定进行。应当能够清楚地鉴别对每位受试者在每个试验周期给予的药品。
8、临床试验方案确定,包括预BE试验方案和正式试验方案。预BE 试验方案:变异系数(个体内变异系数)、受试者数量、生物样本采样量、采样点、采样时间等;正式试验基于预BE确定的个体内变异系数,最终确定受试者数量,以及根据预BE优化修正的生物样本采样量、采样点、采样时间和洗脱期。
变异系数的信息查询:EMEA的药物european public assessment report(EPAR):;FDA药物标签review,?:drugsatfda/,以及PUBMED-NCBI,查询相关药物个体内变异系数RSD
文献。
ANOVA
试验方案设计根据药物特点,可选用1)两制剂、单次给药、交叉试验;2)两制剂、单次给药、平行试验;3)重复试验设计。对于一般药
物选用第1种试验设计,纳入健康志愿者参与研究,每位受试者依照随机顺序接受受试制剂和参比制剂。对于半衰期较长的药物,可选择第2种试验设计,即每个制剂分别在具有相似人口学特征的两组受试者中进行试验。第3种试验设计(重复试验设计)是前两种的备选方案,是指将同一制剂重复给予同一受试者,可设计为部分重复(单制剂重复,即三周期)或完全重复(两制剂均重复,即四周期)。重复试验设计适用于部分高变异药物(个体内变异≥30%),优势在于可以入选较少数量的受试者进行试验。
9、伦理委员会审查资料:
(1)受试样品质量标准及检验报告
(2)参比制剂质量标准及检验报告
(3)药物临床试验批件
(4)伦理审查申请表
(5)临床试验方案,摘要、说明
(6)研究者简历、研究者手册
(7)受试样品和参比制剂说明书
(8)知情同意书
10、将合格的试制样品和参比制剂递交给临床试验机构,该过程注意在GMP和GCP条件下进行,应符合各项SOP标准。
11、CRO对临床试验机构相关人员进行GCP、研究方案和SOP培训,启动临床研究。
12、招募受试者,根据制定试验方案中相关受试者筛选和排除标准进
行。有对适当比例(约1/3)女性受试者的要求。
13、依照临床试验方案、GCP、SOP等进行给药采血及数据收集。这一阶更应加强CRO所派遣的临床监察员和申办方即我公司所派的监督检查者对临床机构的监督检查。
14、血样运输至分析检测机构(冷链运输——CRO负责),严格执行各种GCP SOP要求。
15、根据之前建立的与本生物样本所适应的方法学进行样本检测。要求:规范性、真实性和可追溯性。
16、对药代参数出具统计分析报告,若具备欧盟或FDA标准,应按照最高标准进行统计分析。
17、统计报告及总结报告完成。应该是受试样品与参比制剂具备生物等效性的报告。
18、三套原始资料、及光盘和报告,此过程应为CRO与申办方即我公司的完成的生物等效试验资料交接。