利用深度学习的核苷酸序列预测分析
核苷酸序列分析
核苷酸序列分析
ORF
Getorf
Plotorf ORF Finder BestORF
基因开放阅读框/基因结构分析识别工具
http://bioweb.pasteur.fr/seqanal/interfaces/getorf.html
http://bioweb.pasteur.fr/seqanal/interfaces/plotorf.html /gorf/gorf.html /all.htm
• GlimerM适于恶性疟原虫、拟南芥、曲霉菌 和水稻 • 对mRNA, cDNA, EST, 宜用GetOrf, ORF Finder, Plotorf, BestORF 等
核苷酸序列分析
ORF
应用ORF Finder预测水稻瘤矮病毒 (RGDV)S8片断的ORF
• ORF Finder: /gorf/gorf.html
核苷酸序列分析
重复序列分析 开放读码框(open reading frame, ORF)的识别 基因结构分析 内含子/外显子剪切位点识别 选择性剪切分析 CpG 岛的识别 核心启动子/转录因子结合位点/转录启始位 点的识别 转录终止信号的预测 GC含量/密码子偏好性分析
核苷酸序列分析
ORF
重复序列分析
Web/Linux
Web Web Web/Linux Linur
FGENESH+ /++
/generation/
r.it/~webgene/genebuilder.html /all.htm /genomescan.html /Software/Wise2/ /grailexp/ /seq-search/genesearch.html
• Kozak规则: ORF中起始密码子ATG前后的碱基具 有特定的偏好性。若将第一个ATG中的碱基分别 标为1、2、3位,则Kozak规则可描述如下:
核酸序列预测分析的基本思路

核酸序列预测分析的基本思路当我们得到一个DNA序列时,一般都需要对该片段进行分析,确定它的功能区域,寻找调控区域、编码区域,预测其编码蛋白,这些就是我们研究DNA序列的目的。
核酸序列的预测就是在核酸序列中寻找基因,找出基因的位置及功能位点,以及标记已知的序列模式等过程。
在此过程中,确认一段DNA序列是一个基因需要有多个证据的支持:1、一般而言,在重复片段频繁出现的区域里,基因编码区和调控区不太可能出现;2、如果某段DNA片段的假想产物与某个已知的蛋白质或其它基因的产物具有较高序列相似性的话,那么这个DNA片段就非常可能属于外显子片段;3、在一段DNA序列上出现统计上的规律性,即所谓的“密码子偏好性”,也是说明这段DNA是蛋白质编码区的有力证据;4、其它的证据包括与“模板”序列的模式相匹配、简单序列模式如TATA Box等相匹配等。
一般而言,确定基因的位置和结构需要多个方法综合运用,而且需要遵循一定的规则:1、对于真核生物序列,在进行预测之前先要进行重复序列分析,把重复序列标记出来并除去;2、选用预测分析程序时要注意程序的物种特异性,要弄清程序适用的是基因组序列还是cDNA序列;很多程序对序列长度也有要求,有的程序只适用于长序列,而对EST这类残缺的序列则不适用。
要注意的是,尽管各种预测方法都基于现有的生物学数据和已有的生物学知识,但在不同模型或算法基础上建立的不同分析程序有其一定的适用范围和相应的限制条件,因此最好对同一个生物学问题尽量多用几种分析程序,综合分析各种方法得到的结果和结果的可靠性。
此外,生物信息学的分析只是为生物学研究提供参考,这些信息能提高研究的效率或提供研究的思路,但很多问题还需要通过实验的方法得到验证。
一般地,核酸序列信息分析的基本思路:编码区序列(简称CDS)与EST数据比较→寻找感兴趣ESTS (标准:长度≥100bp,同源性介于50%~85%之间)→所选ESTs与GenEmble数据库比较→找出未克隆ESTs→再与dbEST、dsSTS、dbHTGs、MGD及UniGene数据库比较搜寻重叠群Contigs→设计引物进行PCR扩增或筛选cDNA文库或索取cDNA克隆号进行电子拼接获取全长cDNA→基因定位、表达、结构、功能检测分析等。
核苷酸分析报告

核苷酸分析报告报告书尊敬的贵公司:非常感谢您选择我们公司进行核苷酸分析,并且感谢您对我们的信任和支持。
现在,我们给您呈上此次的核苷酸分析报告。
一、分析结果通过我们公司的高精度核苷酸分析设备,经过严格的实验步骤和科学的数据处理,最终得到以下结果:1. 样品名称: A12345。
2. 样品来源:贵公司,送样人:张三,样品接收时间:2021年6月1日。
3. 核苷酸含量:样品A12345中核苷酸的总含量为0.25mg/g,其中A、C、G、T四种核苷酸分别为0.03mg/g、0.06mg/g、0.08mg/g、0.08mg/g。
二、分析方法和标准本次核苷酸分析采用的是高效液相色谱法(HPLC)进行的。
该方法具有精度高、重现性好、分析速度快等特点,同时符合国家相关标准和规定。
三、分析结论及建议通过核苷酸分析结果,我们可以得出以下结论:经分析,样品A12345中核苷酸的总含量为0.25mg/g,其中A、C、G、T四种核苷酸分别为0.03mg/g、0.06mg/g、0.08mg/g、0.08mg/g。
按照国家相关标准和规定,该样品中核苷酸含量处于中等水平。
针对上述分析结果,我们建议您在后续的研究和开发过程中,结合其他指标和要求,综合考虑,做出科学的、合理的研发计划和生产方案。
四、感谢信在此,我们再次感谢贵公司的信任和支持。
如有任何问题,欢迎随时与我们联系。
我们将竭诚为您服务!此致敬礼!xxxx年xx月xx日xx公司签章:xxxx附:核苷酸分析报告原始数据(仅供参考)。
基于深度学习的DNA序列分析与分类

基于深度学习的DNA序列分析与分类基于深度学习的DNA序列分析与分类DNA序列是生物学研究中至关重要的一种数据形式。
通过对DNA序列的分析和分类,可以揭示生物之间的进化关系、基因功能以及遗传疾病的发生机制等。
而深度学习作为一种强大的机器学习方法,已经在许多领域展现出了非凡的能力。
本文将探讨基于深度学习的DNA序列分析与分类方法,以及其在生物学研究中的应用。
首先,深度学习的核心思想是通过多层神经网络模型来学习数据的特征表示。
在DNA序列分析中,深度学习可以通过学习DNA序列中的局部特征和全局特征,实现对DNA序列的分类和预测。
在DNA序列分析中,最常见的任务之一是基因识别。
基因识别是指从DNA序列中确定编码蛋白质的基因区域。
深度学习可以通过训练一个嵌套的卷积神经网络(CNN)模型,从DNA序列中提取特征,并判断每个碱基是否属于基因区域。
通过这种方法,深度学习可以有效地识别出基因区域,进而推断基因的功能和表达水平。
另一个重要的DNA序列分析任务是DNA突变的预测。
DNA突变是指DNA序列中的碱基发生变异,可能导致遗传疾病和肿瘤的发生。
深度学习可以通过学习DNA序列中的模式和规律,预测潜在的DNA突变。
例如,可以使用循环神经网络(RNN)模型来建模DNA序列中的时序信息,进而预测基因突变的发生概率。
这种方法可以帮助研究人员更好地理解DNA突变的机制,并为疾病的早期预测和诊断提供有力支持。
此外,深度学习还可以用于DNA序列的分类和聚类分析。
通过训练一个深度神经网络模型,可以将不同类型的DNA序列进行分类,从而揭示不同物种之间的进化关系和遗传变异。
此外,通过将DNA序列映射到一个低维空间,可以使用深度学习模型进行DNA序列的聚类分析,从而发现隐藏在大规模DNA序列数据中的模式和共同特征。
综上所述,基于深度学习的DNA序列分析与分类方法在生物学研究中具有广泛的应用前景。
通过深度学习的强大特征学习能力,可以从DNA序列中挖掘出更多的信息和知识,为生物学研究提供更精确、高效的工具和方法。
第五章对核酸序列进行预测分析

关于假基因的来源一般认为是由mRNA反转录成cDNA,然后整合在基因
组中。假基因同cDNA一样没有内含子序列,也没有启动基因转录的启动子 序列,而在5’端都有mRNA分子特有的多聚腺苷[poly(A)]序列。
由于假基因没有生物学功能,所以不再受到进化的选择压力,因此在
假基因中可以积累许多突变,并常常同时存在三种终止密码子序列。假基 因是由功能基因演变而来,可以看作是进化的一种遗迹。
为什么RNA聚合酶能够仅在启动子处 结合呢?
• 启动子处的核苷酸顺序具有特异的形状以便与RNA聚合酶结 合,就好像酶与其底物的结构相恰恰适合一样。将100个以 上启动子的顺序进行了比较,发现在RNA合成开始位点的上 游大约10bp和35bp处有两个共同的顺序,称为-10和-35序 列。 • 共同序列: -10 TTGACATATATT 原核生物 Pribnow盒 -35
-70-80bp
AATGTGTGGAAT 真核生物 TATA盒
GCCTCAATCT 真核生物 CAAT盒
• 生物中有许多启动子,如大肠杆菌约有2000个启动子。各启 动子的效率可不相同,大肠杆菌的强启动子每2秒钟启动一次 转录,而弱启动子每10分钟才启动一次
为什么要分析预测启动子
• 是否使启动子序列改变 • 什么与启动子结合 • 调控基因表达
真核生物启动子
一个真核基因按功能可分为两部分,即调节区和结构基因。结构基因的DNA序列指 导RNA转录;如果该DNA序列转录产物为mRNA,则最终翻译为蛋白质。调节区由两类元 件组成,一类元件决定基因的基础表达,又称为启动子;另一类元件决定组织特异性 表达或对外环境及刺激应答;两者共同调节表达。 • RNA聚合酶Ⅱ识别的启动子与原核生物的启动子相似,也具有两个高度保守的共有 序列。其一是在-25附近的一段AT富集序列,其共有序列是TATAA,称为TATA盒。 TATA盒与原核的Pribonow盒相似,是转录因子与DNA分子结合的部位。其二是在多数 启动子中,-70附近共有序列CAAT区,称为CAAT盒。除以上两个区域外,有些启动子 上游中含有GC盒,此GC盒与CAAT盒多位于-40~110之间,它们可影响转录起始的频率。 另外,有少量基因缺乏TATA盒,而由起始序列(Inr)与RNA聚合酶Ⅱ直接作用启动基 础转录的开始。启动子决定了被转录基因的启动频率与精确性,同时启动子在DNA序 列中的位置和方向是严格固定的,是由5′到3′方向。
生物信息学-第五章-核苷酸序列分析

Web/Windows/ Linux
Web/Windows/ Linux
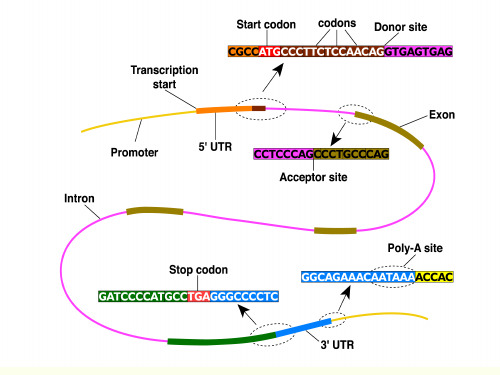
基因结构分析
剪切位点识别:NetGene2
http://www.cbs.dtu.dk/services/NetGene2/
Байду номын сангаас
基因结构分析
NetGene2输出结果
供体位点 可信度
受体位点
基因结构分析
mRNA剪切位点识别:Spidey
可同时输入多条cDNA/mRNA序列与同一条基因组序列进行分析
输入基因组序列 或序列数据库号
判断用于分析的序列间的差异, 并调整比对参数 比对阈值 选择物种
输入mRNA.txt文档中的 6条序列
不受默认内含子长度限制, 默认长度:内部内含子 为35kb, 末端内含子为100kb 输出格式
基因结构分析
/spidey
• NCBI开发的在线预测程序 • 用于mRNA序列同基因组序列比对分析
基因结构分析
Spidey序列提交页面
序列在线提交形式:
界面中有两个窗口:
• 上方窗口用于输入基因组序列(直接粘贴序列或用Genbank ID/AC号) • 下方窗口用于输入cDNA/mRNA序列(直接粘贴序列或用Genbank ID/AC号)
第三章核苷酸序列分析基因组序列cdna序列编码区预测codonbiasgccontent限制性酶切位点基因结构分析选择性剪切转录调控因子序列比对功能注释kegggo系统发育树蛋白质序列翻译蛋白质理化性质二级结构预测结构域分析重要信号位点分析三级结构预测基因组功能分析核苷酸序列分析基因预测开放读码框genscangenomescanglimmer基因结构分析内含子外显子剪切位点netgene2spidey选择性剪切prosplicerspidey转录调控序列分析启动子转录起始位点epdcistercpg岛cpgplot转录终止信号hcpolya序列组分分析gc含量genskew限制性核酸内切酶位点nebcutter密码子偏好性使用codonw开放读码框的识别?开放读码框openreadingframeorf是一段起始密码子和终止密码子之间的碱基序列?orf是潜在的蛋白质编码区whatdoesthissequencemean
深度学习在基因序列预测中的应用剖析
深度学习在基因序列预测中的应用剖析随着科技的迅猛发展和生物学研究的深入,基因组学成为了生命科学中一个重要的研究领域。
基因序列预测是基因组学中不可或缺的一个任务,它旨在准确且高效地确定一个生物体基因组中的基因位置和功能。
近年来,深度学习技术的快速发展为基因序列预测带来了新的机会和挑战。
深度学习是一种机器学习的方法,它通过构建多层神经网络模型,从大规模数据中提取特征并进行预测。
与传统基因序列预测方法相比,深度学习技术具备以下几个优势。
首先,深度学习可以自动学习特征表示。
传统的基因序列预测方法需要手动构建特征提取器,而深度学习可以通过反向传播算法自动学习具有良好判别能力的特征表示。
这使得深度学习模型能够更好地适应不同类型的基因序列数据。
其次,深度学习可以处理大规模复杂的基因组数据。
随着高通量测序技术的普及,研究人员可以轻松获得大量的基因序列数据。
深度学习模型具备处理大规模数据的能力,能够更好地发现数据中的模式和规律。
另外,深度学习模型具备良好的泛化能力。
泛化能力是指模型在未见过的数据上进行预测的能力。
基因序列预测任务需要面对不同生物种类、不同基因特征以及不同噪声水平的挑战,传统机器学习方法的泛化能力较弱。
而深度学习模型可以通过大量训练数据的学习,提高对未知数据的预测能力。
在基因序列预测中,深度学习技术已经被广泛应用于多个子任务中。
首先,深度学习在基因定位预测中取得了重要进展。
基因组中的基因定位信息对于基因功能研究至关重要。
传统的基因定位预测方法主要基于序列特征、组学特征等进行模型训练和预测。
然而,这些方法对于复杂的基因组数据的建模能力有限。
深度学习模型通过多层网络结构和自动学习特征表示的能力,可以更好地捕捉基因组中的模式和特征,从而提高基因定位预测的准确性。
其次,深度学习在基因结构预测中也取得了显著进展。
基因结构预测旨在识别基因组中的外显子、内含子和剪接位点等结构信息。
传统的基因结构预测方法主要基于统计模型、特征提取和机器学习进行,但面对复杂的基因组结构和多样性基因结构的挑战时,表现相对有限。
基于深度学习的核小体位点预测方法
/引 言
核小体是由D N A 和组蛋白形成的染色质基本结构单 位 。每个核小体由14 6 b p 的 D N A 缠绕组蛋白八聚体近两 圈形成,核小体核心颗粒之间通过60 b p 左右的连接D N A 相连[1]。核小体定位在基因表达、D N A 复制、D N A 修复 和 R N A 剪切等细胞活动起着重要的作用,同时组蛋白修饰 调节异常在重大疾病中的作用与核小体位置信息也有着直 接的联系,因此研究核小体在D N A 序列上的位点预测方法
2019 年 3 月 第 40卷 第 3 期
计算机工程与设计
C O M PU TER EN G IN EER IN G AN D D ESIG N
M ar. 2019 V ol. 40 No. 3
基于深度学习的核小体位点预测方法
钱慎一 , 李代祎, 王 晓 , 刘慧慧
( 郑 州 轻 工 业 学 院 计 算 机 与 通 信 工 程 学 院 ,河 南 郑 州 450000) 摘 要 :为实现在海量的被测序D N A 序列中快速、准确的定位核小体,解决传统人工实验法和被提出的一些计算方法耗 时长和准确率低等问题,迫切需要设计一种快速有效的核小体自动化定位方法。在 基 于 伪 核 苷 酸 K -联体特征提取的基础上 构造样本集的特征向量,提 出 在 T e n s o r F lo w 框 架 下 利 用 卷 积 神 经 网 络 (C N N ) 构建核 小 体 定 位 的 网 络 预 测 模 型 。在预测 模型上分别对智人、线 虫 和 果蝇 3 个基准数据集进行 交 叉 验 证 测 试 ,预 测 准 确 率 分 别 为 8 8 . 2 1 % 、89. 19f 、85. 0 7 % , 实 验 结 果 表 明 ,该 预 测 模 型 性 能 高 于 目 前 已 有 预 测 模 型 。 关 键 词$ 核小体位点;向量化;特征提取;卷积神经网络& 交叉验证 中图法分类号!T P 309 文献标识号:A 文 章 编 号 $ 1000-7024 (2019) 03-0862-07 do i : 10. 16208!. is s n l000-7024. 2019. 03. 044
生物信息学中的基因序列分析与预测方法解析
生物信息学中的基因序列分析与预测方法解析生物信息学是将计算机科学和生物学相结合,以研究生物信息的存储、管理、分析和应用为主要内容的学科领域。
基因序列是生物体内决定遗传特征的重要信息之一,其分析与预测方法在生物信息学研究中具有重要的作用。
本文将对基因序列分析与预测方法进行详细解析。
基因序列分析是指对DNA序列进行处理、解读和研究的过程。
主要方法包括序列比对、序列注释、序列聚类和序列可视化等。
首先是序列比对(Sequence Alignment)。
序列比对是将两个或多个序列进行对比,找出它们之间的相似性和差异性。
常用的比对算法包括全局比对、局部比对和多序列比对。
全局比对方法常用于相对较短的序列,如Smith-Waterman算法和Needleman-Wunsch算法。
局部比对方法则适用于比对长序列或序列的局部区域,如BLAST算法和FASTA算法。
多序列比对则是比对超过两个的序列,如CLUSTALW和MUSCLE等方法。
其次是序列注释(Sequence Annotation)。
序列注释是指对DNA或蛋白质序列进行对应功能、结构和进化信息的标注。
常见的注释信息包括基因识别、编码区域和非编码区域的注释、启动子和终止子的预测、外显子和内含子的划分等。
常用的注释软件有NCBI的ORFfinder、Genscan、GeneMark和Ensembl等。
序列聚类(Sequence Clustering)是将具有相似特征的序列归类到同一群集中的过程。
聚类方法可以将大量的生物序列整合到一起,发现其共同的特征和模式。
聚类方法包括基于序列相似性的聚类和无监督聚类方法。
常用的聚类算法包括K-means算法、自组织映射(SOM)和层次聚类等。
序列可视化(Sequence Visualization)是通过图形化的方式展示序列的特征和模式。
常见的可视化方法包括序列Logo的绘制、热图和网络图的构建等。
序列Logo是通过将相同位置上不同碱基或氨基酸的频率进行比较,生成一个图形化的显示,用于研究序列中的保守性和突变等信息。
基于深度学习的DNA序列分析技术研究
基于深度学习的DNA序列分析技术研究一、引言随着基因测序技术的不断发展和普及,大量的DNA序列数据得以产生并储存,如何高效地进行数据分析和生物信息学研究成为了当前的热点问题。
传统的生物信息学方法往往依赖于手动设计特征和建模,需要人工参与且效率低下,也难以应对大规模的序列数据处理。
而深度学习作为一种自动化的机器学习方法,提供了新的解决方案和技术支持。
本文将详细介绍基于深度学习的DNA序列分析技术研究进展和应用前景。
二、基础知识2.1 DNA 序列DNA(脱氧核糖核酸)是生物体遗传物质的基础,是由四种不同的碱基(腺嘌呤 A,胸腺嘧啶 T,鸟嘌呤 G 和胞嘧啶 C)按照一定的顺序组成的长链分子。
每三个连续的碱基构成一个密码子,指定一个氨基酸或起始、终止的信号。
整个 DNA 序列决定了生物个体的自然特征和生理、病理过程。
2.2 深度学习深度学习是一种基于神经网络的机器学习技术,其核心是构建和训练多个层次的神经网络模型,实现从数据中学习表征和特征,然后再利用这些表征和特征进行分类、预测或者生成等运算。
三、基于深度学习的 DNA 序列分析技术3.1 基因识别基因识别是生物信息学的一个关键任务,其目的在于识别出DNA 中的基因编码区域。
传统的基因识别方法往往依赖于人工定义的规则和特征,如开放阅读框架(ORF)长度、启动子、终止子等,这些方法存在着灵敏度低、精度不高和易受到干扰等问题。
基于深度学习的基因识别方法可以将 DNA 序列直接输入模型,自动学习重要的特征和规则,大大提高了识别效率和准确性。
3.2 基因功能预测基因的生物学功能包括编码蛋白质和非编码 RNA,参与细胞代谢和信号传导等生命过程。
基于深度学习的基因功能预测可以进一步挖掘 DNA 序列中的信息,如序列保守性、启动子、结构域等,以预测基因的生物学功能。
这些预测结果对于基因工程、生物医学研究和新药开发等领域具有重要的指导意义。
3.3 基因分类与聚类基因分类和聚类是生物信息学研究中的常见任务。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
利用深度学习的核苷酸序列预测分析第一章:引言
核苷酸序列预测分析是生物信息学领域中重要的研究方向之一。
这一领域的研究目的在于寻找生物体内的一些重要特征或者预测
生物体的功能。
在过去的几十年中,传统的研究方法主要使用了
手动提取特征。
这种方法虽然经过了长期的研究,但是由于人工
提取特征的难度和工作量大,容易出错,所以导致许多问题。
而
现在随着深度学习算法的出现和生物学数据量的不断增大,利用
深度学习对核苷酸序列进行预测分析成为了一种新的选择。
深度
学习算法可以自动从原始数据中学习特征,从而提高预测的准确
率和效率。
因此,利用深度学习的核苷酸序列预测分析已经吸引
了广泛关注。
第二章:深度学习在核苷酸序列预测分析中的应用
深度学习能够自动提取特征,适用于各种生物信息学的任务,
包括分子序列分类、序列注释、构象分析和功能预测等。
在核苷
酸序列预测分析方面,深度学习方法主要应用于下列任务:DNA
序列特征提取、RNA序列特征提取、基因结构预测、外显子预测、编码RNA预测和蛋白质结构预测。
2.1 DNA序列特征提取
DNA序列特征提取是指从DNA序列中提取有意义信息的过程。
深度学习方法可以从原始DNA序列中提取出单核苷酸、二核苷酸
和三核苷酸等信息。
经过堆叠、卷积和池化等处理,可以自动地
提取出有意义的生物信息。
将提取出的特征输入到分类器中,可
以获得更好的分类效果。
2.2 RNA序列特征提取
RNA序列特征提取是指从RNA序列中提取有意义信息的过程。
深度学习方法可以从原始RNA序列中提取出多个序列特征,包括
稀疏特征、稠密特征和非线性特征等。
这些特征可以用来推断
RNA结构和RNA生物功能。
2.3 基因结构预测
基因结构预测是指预测基因序列中的外显子和内含子区域。
深
度学习方法可以通过学习对其进行预测。
其可以从原始DNA序列
中自动学习特征,利用深度学习算法进行分类,从而预测基因结构。
2.4 外显子预测
外显子预测是指从未知的核苷酸序列中预测出外显子序列。
深
度学习方法可以使用多种架构,如卷积神经网络和循环神经网络等。
该方法可以提高外显子预测的准确性和效率。
2.5 编码RNA预测
编码RNA预测是指预测RNA是否具有编码蛋白质的能力。
深
度学习方法可以自动提取原始RNA序列的特征,这些特征可以用
于推断RNA是否具有编码蛋白质的能力。
基于深度学习的RNA
编码能力预测方法在真实数据集上获得了非常好的结果。
2.6 蛋白质结构预测
蛋白质结构预测是指通过预测氨基酸序列的三维结构来预测蛋
白质的结构。
深度学习方法可以用于从氨基酸序列中提取特征,
预测蛋白质的结构。
此外,深度学习方法还可以用于预测蛋白质
的相互作用、蛋白质折叠动力学以及蛋白质疾病相关性分析。
第三章:深度学习方法的优缺点
使用深度学习方法在核苷酸序列预测分析中具有许多优点。
这
些优点包括以下几个方面:
- 深度学习算法非常灵活,可以处理不同类型和规模的数据。
- 与传统的方法相比,在许多生物学应用中表现出更高的准确
性和精确度。
- 深度学习模型可自动适应新的数据,无需手动提取特征。
但是,深度学习方法在应用到生物信息学领域中也存在一些缺点:
- 需要运行在高性能计算机上,且需要大量的计算资源和时间。
- 对数据的需求较高。
深度学习方法适合于处理规模较大的数
据集,而对于小规模数据的预测效果并不够优秀。
- 当数据量不够充分或者质量较差时,模型可能会出现过拟合。
第四章:结论
总之,深度学习方法已经成为生物信息学研究中重要的工具之一。
深度学习方法已经在核苷酸序列预测分析中取得了其研究逐
步成熟的结果,并成功地用于许多生物学应用的实际情况中。
从
实际带来的优点来说,深度学习方法是一个high-level的建模方法,它可无需任何领域专业知识,快速地从海量的数据中精准地学习
到模式,并做出预测或甚至图形化表达。
因此,在生物信息学的
大数据时代,深度学习技术必将得到更广泛的应用。