第四章-DNA序列分析
生物信息学方法在特定基因调控区识别和分析中的应用

生物信息学方法在特定基因调控区识别和分析中的应用随着生命科学的发展,生物信息学方法在分子生物学研究中扮演着越来越重要的角色。
特定基因调控区(gene regulatory region)是指位于基因的上游或下游区域,包含了各种调控元件和转录因子结合位点,是基因表达调控的重要部分。
通过识别和分析这些调控区,可以深入了解基因表达的调控机制,并有助于发现新的治疗和预防疾病的方法。
本文将介绍生物信息学方法在特定基因调控区识别和分析中的应用。
1. DNA序列分析DNA序列分析是指对调控区DNA序列进行计算机处理,以识别其中包含的调控元件和转录因子结合位点,并预测它们对基因表达的影响。
这一过程可以借助许多生物信息学工具实现,如MEME和Weeder等。
这些工具可以进行模式识别和序列比较,从而发现DNA序列中的共同模式和保守序列。
2. ATAC-Seq技术ATAC-Seq技术是一种基于开放染色质的测序方法,用于研究特定细胞类型中基因调控区的开放度。
该技术可以利用转座酶插入开放染色质区域,然后通过PCR扩增和测序来分析这些区域的DNA序列。
通过露出的DNA序列,可以确定基因调控区的开放状态,并预测转录因子的结合位点。
3. CHIP-Seq技术CHIP-Seq技术是一种高通量测序方法,用于鉴定某种转录因子与调控区DNA 结合的位点及其相应的上游基因。
该技术利用可特异地识别转录因子的抗体,将与之结合的DNA序列片段分离出来,并通过测序来鉴定所结合的基因区域。
通过CHIP-Seq技术可以全面地鉴定基因的上游区域和下游区域中的转录因子结合位点,从而为研究基因调控提供基础数据。
4. Hi-C技术Hi-C技术是一种全基因组3D染色质拓扑结构的测序方法,可以用于分析基因调控区的空间结构和相互作用。
通过该方法,可以同时测定两个DNA序列片段之间的空间距离和它们之间的相互作用,从而构建基因组范围的联系图。
利用这一联系图,可以了解基因调控区在三维空间中的位置及其与其他基因区域的互动,从而发现新的调控元件。
新教材 人教版高中生物必修2 第四章 基因的表达 知识点考点重点难点提炼汇总
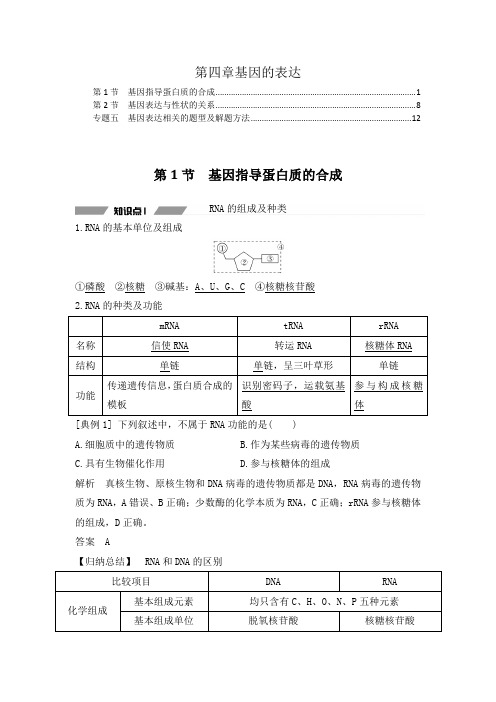
第四章基因的表达第1节 基因指导蛋白质的合成 ........................................................................................... 1 第2节 基因表达与性状的关系 ........................................................................................... 8 专题五 基因表达相关的题型及解题方法 . (12)第1节 基因指导蛋白质的合成RNA 的组成及种类1.RNA 的基本单位及组成①磷酸 ②核糖 ③碱基:A 、U 、G 、C ④核糖核苷酸 2.RNA 的种类及功能 mRNA tRNA rRNA 名称 信使RNA 转运RNA 核糖体RNA 结构 单链单链,呈三叶草形单链功能传递遗传信息,蛋白质合成的模板识别密码子,运载氨基酸参与构成核糖体[典例1] 下列叙述中,不属于RNA 功能的是( ) A.细胞质中的遗传物质 B.作为某些病毒的遗传物质 C.具有生物催化作用D.参与核糖体的组成解析 真核生物、原核生物和DNA 病毒的遗传物质都是DNA ,RNA 病毒的遗传物质为RNA ,A 错误、B 正确;少数酶的化学本质为RNA ,C 正确;rRNA 参与核糖体的组成,D 正确。
答案 A【归纳总结】 RNA 和DNA 的区别比较项目DNARNA化学组成基本组成元素 均只含有C 、H 、O 、N 、P 五种元素 基本组成单位脱氧核苷酸核糖核苷酸碱基A、G、C、T A、G、C、U五碳糖脱氧核糖核糖无机酸磷酸磷酸空间结构规则的双螺旋结构通常呈单链结构【归纳】DNA与RNA的判定方法(1)根据五碳糖种类判定:若核酸分子中含核糖,一定为RNA;含脱氧核糖,一定为DNA。
(2)根据含氮碱基判定:含T的核酸一定是DNA;含U的核酸一定是RNA。
DNA序列的图形表示及其相似性分析

分子生物学:DNA复制

(CsCl gradient centrifuge)
N15
DNA
N14
Semi-Conservation Replication
Source:M. Meselson and F. W. Stahl, Sciences 44:675, 1958.
半半保保留留复复制制-小结
DNA生物合成时,母链DNA解开为两股单链,各自作为 模板(template)按碱基配对规律,合成与模板互补的子链。子代 细胞的DNA,一股单链从亲代完整地接受过来,另一股单链则 完全重新合成。两个子细胞的DNA都和亲代DNA碱基序列一致。 这种复制方式称为半保留复制。
RNA引物的形成
DNA链合成及延长
复制的终止
• RNApol (RNA polymerase)
[Rif S ]
完成对先导链引物的合成
实现DNA复制的转录激活起始
起
• dnaG (primase) [Rif R]
始
完成对后随链引物的合成
较先导链的启动落后一个Okazaki片断
• 完成10±NtRNA引物合成后.
遗传物质的基本属性:基因的自我复制 基因的突变 控制性状的表达
DNA复制
亲代双链DNA分子在DNA聚合酶的作用下, 分别以每 条 单链DNA分子为模板,聚合与 自身碱基可以互补配对的游离的dNTP,合 成出两条与亲代DNA分子完全相同的子代 DNA分子的过程。 主 要 包 括 引 发 、 延 伸 、 终止三个阶段。
复制发动温度敏感突变型(慢停突变) 42℃不能发动DNA复制、但可完成DNA延伸
37 ℃, 5 ci / mM H3-T , 6min
37 ℃, 52 ci / mM H3-T , 6min
基因组学

名词解释:第一章基因组遗传图(连锁图):指基因或DNA标记在染色体上的相对位置与遗传距离。
单位是厘摩cM (基因或DNA片段在染色体交换过程中分离的频率)。
物理图:以已知核苷酸序列的DNA片段(序列标签位点,sequence-tagged site, STS)为“路标”,以碱基对作为基本测量单位(图距)的基因组图。
转录图:以EST(expressed sequence tag ,表达序列标签)为标记,根据转录顺序的位置和距离绘制的图谱。
EST:通过从cDNA文库中随机挑选的克隆进行测序所获得的部分cDNA的5'或3'端序列称为表达序列标签(EST),一般长300-500 bp左右。
序列图(分子水平的物理图):序列图是指整个人类基因组的核苷酸序列图,也是最详尽的物理图。
既包括可转录序列,也包括非转录序列,是转录序列、调节序列和功能未知序列的总和。
基因:合成有功能的蛋白质或RNA所必需的全部DNA序列,即一个基因不仅包括编码蛋白质或RNA的核酸序列,还应包括为保证转录所必需的调控序列。
基因组(genome):生物所具有的携带遗传信息的遗传物质的总和。
基因组学(genomics):涉及基因组作图、测序和整个基因组功能分析的一门学科。
C值:单倍体基因组的DNA总量,一个特定种属具有特征C值C值矛盾(C value paradox):指一个有机体的C值和其编码能力缺乏相关性。
单一序列:基因组中单拷贝的DNA序列。
重复序列:基因组中多拷贝的DNA序列。
复杂性(complexity):基因组中不同序列的DNA总长。
高度重复序列(highly repetitive sequence):重复片段的长度单位在几个到几百个碱基对(base pair,bp)之间(一般不超过200 bp),串联重复频率很高(可达106以上),高度重复后形成的这类重复顺序称为高度重复顺序。
中度重复序列(intermediate repetitive sequence ):重复长度300~7000 bp不等,重复次数在102~105左右。
4第四章遗传物质-基因和染色体

第四章遗传物质——基因和染色体第一节核被膜与核孔复合体细胞核的结构:在固定和染色的细胞中,可观察到细胞有下列结构:核被膜、染色质、核仁、核液(质)四部分。
一、核被膜(nuclear envelope)亦称核膜(nuclear membrane),由此使遗传物质DNA与细胞质分开。
电镜下证实为双层单位膜呈同心性排列。
除两膜之间有间隙外,膜上还有些特化结构。
所以,认为核被膜含义深刻,包括内容多,并执行重要的生理功能。
(一)核被膜结构1 外层核被膜(ONE)(外核膜)膜厚 6.5—7.5nm,相邻细胞质的一面常有核糖体附着,并有时与内质网(RER)相连,因此显得粗糙不平。
2 内层核被膜(INE)(内核膜):膜厚度基本同ONE,膜上无核糖体附着,显得比ONE 平滑。
但在其内表面常附有酸性蛋白质分子的聚合物组成的纤维网状结构(密电子物质),称纤维层(fibrous Lamina)或核纤层(nuclear lamina),又有内致密层之称。
其厚度约在10—20nm(30—160nm),是位于细胞内核膜下的纤维蛋白或纤维蛋白网络。
3 核周隙(perinuclear space)又有核围腔或核围池之称。
指两膜之间的空隙,宽约20—40nm(10—50nm),内充满液态无定形物质(蛋白质、酶类、脂蛋白、分泌蛋白、组蛋白等),它是核质之间活跃的物质交换渠道(有些部位直接与ER或Golgi池相通)。
4 核孔(nuclear pore)核膜并不完全连续,在许多部位,核膜内外两层常彼此融合,形成环状孔道,称为核孔,它们是核质之间的重要通道。
(二)核被膜的主要功能核孔复合体可以看作是一种特殊的跨膜运输蛋白复合体,并且是一个双功能、双向性的亲水性核质交换通道。
双功能表现在它有两种运输方式:被动扩散与主动运输;双向性表现在既介导蛋白质的入核转运,又介导RNA、核糖核蛋白颗粒(RNP)的出核转运。
1、构成核、质之间的天然选择性屏障避免生命活动的彼此干扰,保护DNA不受细胞骨架运动所产生的机械力的损伤2、核质之间的物质交换与信息交流1)通过核孔复合体的被动扩散——小分子物质的转运:核孔复合体作为被动扩散的亲水通道,其有效直径为9~10nm,有的可达12.5nm,即离子、小分子(相对分子质量在60KD以下)以及直径在10nm以下的物质原则上可以自由通过。
bioxm使用说明
(domain);
第四章 DNA与蛋白质序列分析
第一节 序列比对
第二节 Blast应用
第三节 序列功能分析
Question1:
1. 我刚刚分离一个水稻基因片段序列,大概250bp, 我想初步分析一下它是什么基因,编码什么产物以 及是否已经被别人克隆,应该采用什么工具和数据 库? A. Blastn E. blastx B.Blastp F. nr C.tblastn, D.tblastx,
酶切位点分析(载体构建)
基因结构分析/启动子序列分析
Part 1. 初级序列分析
序列的组成/分子量/等电点分析
/
点击“BioXM version 2.6 ” 点击“运行”进行安装
序列组成分析
序列组成分析
序列组成分析
A/G/C/T的组成,尤其是G+C含量的预测(进化?探针设计?)
/Blast.cgi
具体步骤
1.登陆blast主页
/BLAST/ 2.根据数据类型,选择合适的程序 3.填写表单信息 4.提交任务 5.查看和分析结果
第3节 序列功能分析的内容
序列组成/分子量/等电点---初级分析
Part 3. 基因结构分析/启动子序列分析
Genomic DNA 1)基因结构分析: cDNA
第9章_DNA序列分析

第9章_DNA序列分析DNA序列分析是指对DNA序列进行系统性研究和分析的过程。
DNA序列是生物体内的遗传信息的载体,对于了解基因功能、生物演化、疾病发生机制等具有重要意义。
本章将介绍DNA序列分析的方法和应用。
DNA序列分析的方法包括序列比对、基因预测、遗传变异检测和进化分析等。
序列比对是将已知DNA序列与未知序列进行对比,寻找相似之处,从而推断未知序列的功能。
常用的序列比对工具有BLAST、Bowtie等。
基因预测是利用生物信息学方法预测未知DNA序列中的基因位置和功能。
常用的基因预测工具有GeneMark、Glimmer等。
遗传变异检测是通过比较不同个体之间的DNA序列差异,寻找与疾病相关的遗传变异。
进化分析是利用DNA序列比较不同物种之间的遗传差异,推断它们的亲缘关系和演化过程。
常用的进化分析方法有多序列比对、系统发育树构建等。
DNA序列分析在生物学研究和应用领域具有广泛的应用。
在基础研究方面,DNA序列分析可以帮助研究人员了解基因的功能和调控机制。
通过比对不同物种之间的DNA序列,可以揭示物种的进化关系和演化过程。
在医学研究方面,DNA序列分析可以用于疾病的诊断和预测。
通过检测DNA序列中的遗传变异,可以发现与疾病相关的基因突变,并为疾病的治疗和预防提供理论基础。
在农业研究方面,DNA序列分析可以应用于作物和畜禽的遗传改良。
通过分析作物和畜禽的DNA序列,可以挖掘有益基因和导育改良品种,提高农作物和畜禽的产量和品质。
随着高通量测序技术的发展,DNA序列分析在研究领域的应用也得到了大幅度的提升。
高通量测序技术可以快速、准确地获取大量的DNA序列信息,为DNA序列分析提供了更为丰富的数据。
同时,也为DNA序列分析提供了更多的挑战,如序列比对的速度和精度、大规模数据的储存和分析等。
因此,进一步研发和改良DNA序列分析的方法和工具,提高分析效率和准确性,将是今后的研究重点。
综上所述,DNA序列分析是一项重要的生物信息学研究方法,具有广泛的应用前景。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
引言
表达序列标签分析
序列对位排列
4.1 引言
4.1.1 为什么要分析DNA序列
序列及所代表的类群间的系统发育关系 限制性酶切(位点)图谱 通过内含子和外显子(intron/exon)预测所确 定的遗传结构
通过对可读框(open-reading frame, ORF)分析 推导蛋白质编码序列(coding sequence, CDS)
说明两条序列的相似程度 ——〉定量计算
两条序列的相似程度的定量计算 – 相似度,它是两个序列的函数,其值越大,表示 两个序列越相似 – 两个序列之间的距离。距离越大,则两个序列的 相似度就越小
距离计算的不足 字符编辑操作(Edit Operation)
字符编辑操作可将一个序列转化 为一个新序列
5、用于序列相似性打分的权值矩 阵(Weight Matrices)
表3.3 转移矩阵 A 0 5 T 5 0 C 5 1 G 1 5
C
G
0
0
0
0
1
0
0
1
C
G
-4
-4
-4
-4
5
-4
-4
5
C
G
5
1
1
5
0
5
5
0
转移矩阵表
嘌呤(腺嘌呤A,鸟嘌呤G)有两 个环,嘧啶(胞嘧啶C,胸腺嘧啶T) 只有一个环。 转换(transition),如AG、 CT。
Phrap
基于swat算法 使用全序列质量信息 全基因组、EST 通常与Phred和consed联合应用 /phredphra pconsed.html
Phrap
命令及参数 phrap lesson.seq.screen -minmatch 20 -minscore 40 -view -new_ace >phrap.out 结果
– Match(a,a)
– Delete(a,-) – Replace(a,b) – Insert(-,b)
扩展的编辑操作
ACCGACAATATGCATA ATAGGTATAACAGTCA
ACCGACAATATGCATA ACTGACAATATGGATA
不同编辑操作的代价不同
为编辑操作定义函数w,它表示“代价 (cost)”或“权重(weight)”。 对字母表中的任意字符a、b,定义 w (a, a) = 0 w (a, b) = 1 ab w (a, -) = w ( -, b) = 1
也可以使用得分(score)函数 来评价编辑操作
p (a, a) = 2 p (a, b) = -1 a b p (a, -) = w ( -, b) = -1
(ii) 氨基酸突变代价矩阵GCM (iii)疏水矩阵 (iv)PAM矩阵 (v) BLOSUM矩阵
氨基酸突变代价矩阵GCM
GCM(Genetic Code Matrix,Haig and Hurst,1991)
如果变化一个碱基使某些氨基酸的密码子改变为另一些氨基 酸的密码子,其替换代价为1;
对蛋白质的结构和功能不产生太大影响的替换较高。
这些点突变已经被进化所接受。这意味着在进化历 程上相关的蛋白质在某些位置上可以出现不同的氨 基酸。
一个PAM就是一个进化的变异单位,即1%的氨基酸改 变 PAM有一系列的替换矩阵,每个矩阵用于比较具有特 定进化距离的两个序列。例如,PAM120矩阵用于比 较相距120个PAM单位的序列。 一个PAM-N矩阵元素(i,j)的值反应两个相距N个 PAM单位的序列中第i种氨基酸替换第j种氨基酸的频 率。 将PAM1自乘N次,可以得到PAM-N。 Dayhoff等第一次使用了log-odd处理,矩阵中的取代分 值同目标频率与背景频率的比值的自然对数成比例。 Dayhoff等人只发表了PAM250,通常在较高的PAM值 处得到最佳结果,比如在PAM200到250之间,较低值 的PAM矩阵一般使用于高度相似的序列 (Altschul,1991)。
/gorf/gorf.html
4.2 表达序列标签A序列互补的DNA
双链的cDNA插入合适的分析 表达序列标签(expressed sequep),代表特定 组织或发育阶段表达的基因。
第二条序列头尾颠倒
CTAGTCGAGGCAATCT GAACAGCTTCGTTAGT
?
3、通过点矩阵进行序列比较
“矩阵作图法” 或 “对角线作图”
4、 序列的两两比对
序列的两两比对 (Pairwise Sequence Alignment) 按字符位置重组两个序列,使得两个序列 达到一样的长度
lesson.seq.screen.contigs lesson.seq.screen.singlets lesson.seq.screen.view lesson.seq.screen.ace phrap.out
4.3 序列对位排列
序列比较的根本任务是:
发现序列之间的相似性 辨别序列之间的差异
概念: 两个序列s 和 t 的比对代价等于将s 转化为t 所 用的所有编辑操作的代价和 s 和t 的最优比对是所有可能的比对中代价最小 的一个比对 s 和 t 的真实距离应该是在代价函数w值最优时 的距离,记为dw(s,t)。 例如: s: AGCACACA t: ACACACTA cost=2
序列比对的目的是寻找一个代价最小的比对。
(1)核酸打分矩阵设DNA序列所用的字母表为 = { A,C,G,T }
a. 等价矩阵 b. BLAST矩阵 c. 转移矩阵表
表3.1 等价矩阵表 A A T 1 0 T 0 1 C 0 0 G 0 0 A T 表3.2 BLAST矩阵 A 5 -4 T -4 5 C -4 -4 G -4 -4 A T
颠换(transversion),如AC、 AT 转换发生的频率远比颠换高
A A T C G 0 5 5 1 T 5 0 1 5 C 5 1 0 5 G 1 5 5 0
(2)蛋白质打分矩阵
(i)等价矩阵
1 i j Rij 0 i j
其中Rij代表打分矩阵元素 i、j分别代表字母表第i和第j个字符。
PAM1矩阵
A RA N
R N D C Q E G H I L K M F P S T W Y
D
C
Q
E
G
H
I
L
K
M
F
P
S
T
W
PAM250矩阵
A RA N
R N D C Q E G H I L K M F P S T W Y
D
C
Q
E
G
H
I
L
K
M
F
P
S
T
W
BLOSUM矩阵
BLOSUM矩阵是由Henikoff首先提出的另一种 氨基酸替换矩阵(Henikoff,1992),采用与 PAM同样的方式可以建立BLOSUM替换矩阵 BLOSUM矩阵则是从蛋白质序列块(短序列) 比对而推导出来的。 基本数据来源于BLOCKS数据库,其中包括了 局部多重比对(包含较远的相关序列,同在 PAM中使用较近的相关序列相反)。 通过直接观察获得数据而不是通过外推获得。 同PAM模型一样,也有许多编号的BLOSUM矩 阵。
结果
可读框:一个起始密码子(ATG)和终止密码
子( TAA ,TAG,TGA )之间的序列
一般是从DNA序列而非RNA序列来判断可读
框的存在
一个双链的DNA有6个潜在的可读框
原核生物的编码区是一个单独的ORF,真核
生物编码区含有内含子,要分析真核基因的编 码区还必须识别出内含子和外显子的边界,不 过若使用cDNA序列,则问题可大大简化。
目的:
相似序列 相似的结构,相似的功能 判别序列之间的同源性 推测序列之间的进化关系
序列比较的基本操作是比对(Alignment)
– 两个序列的比对是指这两个序列中各个字符
的一种一一对应关系,或字符的对比排列 。
设有两个序列: GACGGATTAG,GATCGGAATAG Alignment1: GACGGATTAG || | GATCGGAATAG Alignment2: GA-CGGATTAG || |||||||| GATCGGAATAG
4.1.2
5’ 5’UTR
基因结构与DNA序列分析
3’ 外显子 内含子
外显子
内含子
外显子
3’UTR
单链基因组DNA
转录
5’UTR mRNA 翻译 蛋白质 CDS 3’UTR
非翻译区:
在DNA和RNA中均有,位于 CDS两侧,在3’端的UTR是高度特异的。 概念性翻译: 六框翻译(six-frame translation)
EST与cDNA的关系
5’
3’
EST CDS UTR
4.2.2 EST数据库: EMBL, GenBank(dbEST)
4.2.3
EST分析
序列相似性查询 序列组装 序列聚类
4.2.4
电子克隆cDNA全长序列
根据大量EST具有相互重叠的性质,由一个查询 序列开始,依靠EST数据库在计算机上对EST进行 两短延伸,从而获得全长的cDNA序列
疏水矩阵
该矩阵是根据氨基酸残基替换前后疏水
性的变化而得到得分矩阵。
若一次氨基酸替换疏水特性不发生太大
的变化,则这种替换得分高,否则替换 得分低。
PAM矩阵
PAM矩阵是建立在进化的点突变模型PAM(Point Accepted Mutation,Dayhoff et al.,1978)基础上。 Dayhoff等研究了71个相关蛋白质家族的1572个突变, 发现氨基酸的替换并不是随机的,一些氨基酸的替 换比其它替换更容易发生