新能源材料研究中的第一性原理计算
材料科学中第一性原理计算方法研究

材料科学中第一性原理计算方法研究近年来,材料科学领域的研究取得了许多重大突破,其中第一性原理计算方法成为材料设计和研究的重要工具之一。
这种方法通过基本的物理原理和数学方程来研究材料的性质和行为,为材料设计和性能优化提供了新的途径。
第一性原理计算方法是基于量子力学的一种计算方法,从第一性原理出发,通过求解薛定谔方程以及其他相关方程来研究材料的性质。
它不依赖于任何经验参数或假设,能够提供对材料的精确描述和准确预测。
第一性原理计算方法的核心是密度泛函理论(Density Functional Theory,简称DFT),它将体系的物理性质与体系中电子的密度联系起来。
根据Kohn-Sham方程,DFT通过对电子的运动方程进行求解,得到体系的基态电子密度。
通过计算得到的电子密度,可以进一步计算出材料的能带结构、电子态密度、态密度、声子谱、磁性及其它性质。
与传统的实验方法相比,第一性原理计算方法具有独特的优势。
首先,它能够提供物理性质的原子尺度描述,可以捕捉到材料内部微观原子结构的信息。
其次,该方法能够计算和预测材料的多种性质,如电子能带结构、晶格常数、弹性性能、热力学性质等,为材料设计和开发提供了重要参考。
此外,第一性原理计算方法可以帮助解释材料性能背后的基本物理机制,揭示材料特性的微观本质。
近年来,随着计算机性能的不断提升和计算方法的进步,第一性原理计算方法在材料科学中的应用得到了广泛拓展。
例如,它在材料的合成、器件的设计和材料的特性优化等方面发挥了重要作用。
通过预测和优化材料的能带结构和电子态密度,可以筛选出具有优异性能的新材料,为新能源、环境友好材料、传感器和光电器件的研发提供重要支持。
此外,第一性原理计算方法还可以帮助优化材料的力学、热力学和电磁性能,提高材料的功能性能。
尽管第一性原理计算方法为材料科学提供了强大的工具和理论基础,但也面临一些挑战。
首先,该方法对计算所需的资源要求较高,需要大量计算时间和计算机内存。
第一性原理计算在新材料研究中的应用
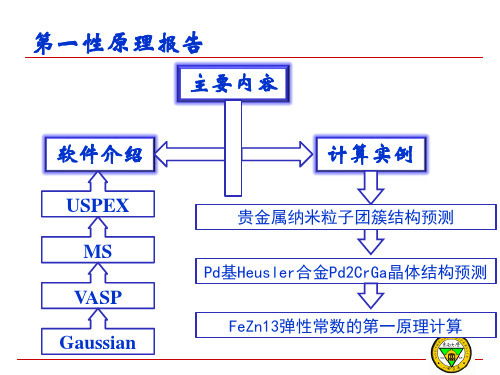
贵金属纳米粒子团簇结构预测
贵金属团簇结构的理论研究
在各种团簇体系中,金团簇体系是目前研究得非常热门的 领域之一。 金原子由于其相对原子质量较大,内层轨道电子的速度可 以与光速相比拟,其4f、5d轨道电子又接近全充满,相对 论效应显著,使其具备很多特殊性质。 大体系金团簇结构以高对称性的富勒烯结构或管状中空笼 结构最为稳定。
VASP
优点
1. 图形界面,操作 方便 2. 建模和结果分析 容易
缺点
1. 在Windows下执 行速度较慢。 2. 可选的计算方法 较少,结果不够 精确
1. 命令行操作,且 Linux系统入门 较难。 2. 建模和结果分析 依赖其他软件。
Castep
Vasp
1. 代码稳定,执行 速度快 2. 可以使用更高级 的计算方法,使 结果更精确
Pd基Heusler合金Pd2CrGa晶体结构预测
态密度
从图中可以看出,Pd2CrGa在两种状态下均表现处很强的自旋极化,而且 在费米面附近尤为明显Cr. 原子的态密度差异是Pd2CrGa总态密度差异的 主要来源,而Pd原子、Ga原子的自旋向上和自旋向下态密度的对称性较 高,对总磁矩的贡献有限. 所以,Cr原子是Pd2CrGa磁性的主要贡献者
离子键团簇 共价键团簇 金属键团簇 (NaCl)n 、(MgO)n 静电作用 Cn 、Sin 、Gen Agn、 Nan 、Cun 共价键结合 电子结合 2-4eV 1-4eV 0.5-3eV
贵金属纳米粒子团簇结构预测
贵金属团簇
由金属Au、Ag、Cu、Pt形成的金属键团簇称为贵金属团簇 近年来,贵金属团簇和纳米颗粒由于其独特的光学、电子 学和催化性能而在生物学、医学、光学、催化和纳米电子 学等领域引起了广泛的兴趣。
第一性原理计算方法在材料科学中的应用

第一性原理计算方法在材料科学中的应用引言:材料科学作为一门跨学科的科学领域,旨在研究材料的性质、结构和性能,以及如何利用这些知识来设计和开发新材料。
而第一性原理计算方法作为一种基于量子力学原理的计算方法,广泛应用于材料科学领域。
本文将介绍第一性原理计算方法在材料科学中的应用,并展示其在材料设计、材料性质预测和材料性能优化等方面的重要性。
一、第一性原理计算方法的基本原理和流程第一性原理计算方法是一种从基本原理出发,仅通过定解问题的边界条件和基本的数学和物理方法,而独立地、直接地得到材料性质的计算方法。
其基本原理是基于薛定谔方程和密度泛函理论,通过求解电子结构和物理性质的基态,来推导和预测材料的性质。
第一性原理计算方法的流程一般包括以下几个步骤:首先,选择适当的计算模型和晶格结构;其次,通过数值方法求解薛定谔方程,得到材料的基态电子密度和能带结构等信息;然后,使用密度泛函理论来计算其他性质,如结构、力学性质、磁性和光学性质等;最后,通过与实验结果对比来验证计算结果的准确性。
二、第一性原理计算方法在材料设计中的应用1. 材料发现和材料库筛选:传统的材料设计通常依赖于试错和实验结果验证的循环迭代,耗费时间和资源。
而第一性原理计算方法能够预测新材料的物理性质,从而加速材料发现过程。
通过计算不同元素和组分的合金化合物,材料科学家可以预测材料的强度、硬度、导电性等重要性能,并筛选出具有潜在应用前景的材料。
2. 材料结构和缺陷研究:材料的结构与其性质密切相关。
通过第一性原理计算方法,可以精确地预测材料的晶体结构、晶格常数、晶粒大小等参数,并探索材料可能存在的结构缺陷和缺陷效应对性能的影响。
这有助于优化材料的结构设计,提高其性能和稳定性。
3. 电子结构和能带计算:材料的电子结构和能带结构对于理解材料的导电性、磁性、光学性质等具有重要意义。
通过第一性原理计算方法,可以准确地计算材料的能带结构、电子态密度分布和费米能级等参数,从而预测材料的导电性、磁性和光学性能。
第一性原理计算简述

第一性原理计算简述第一性原理,英文First Principle,是一个计算物理或计算化学专业名词,广义的第一性原理计算指的是一切基于量子力学原理的计算。
我们知道物质由分子组成,分子由原子组成,原子由原子核和电子组成。
量子力学计算就是根据原子核和电子的相互作用原理去计算分子结构和分子能量(或离子),然后就能计算物质的各种性质。
从头算(ab initio)是狭义的第一性原理计算,它是指不使用经验参数,只用电子质量,光速,质子中子质量等少数实验数据去做量子计算。
但是这个计算很慢,所以就加入一些经验参数,可以大大加快计算速度,当然也会不可避免的牺牲计算结果精度。
那为什么使用“第一性原理”这个字眼呢?据说这是来源于“第一推动力”这个宗教词汇。
第一推动力是牛顿创立的,因为牛顿第一定律说明了物质在不受外力的作用下保持静止或匀速直线运动。
如果宇宙诞生之初万事万物应该是静止的,后来却都在运动,是怎么动起来的呢?牛顿相信这是由于上帝推了一把,并且牛顿晚年致力于神学研究。
现代科学认为宇宙起源于大爆炸,那么大爆炸也是有原因的吧。
所有这些说不清的东西,都归结为宇宙“第一推动力”问题。
科学不相信上帝,我们不清楚“第一推动力”问题只是因为我们科学知识不完善。
第一推动一定由某种原理决定。
这个可以成为“第一原理”。
爱因斯坦晚年致力与“大统一场理论”研究,也是希望找到统概一切物理定律的“第一原理”,可惜,这是当时科学水平所不能及的。
现在也远没有答案。
但是为什么称量子力学计算为第一性原理计算?大概是因为这种计算能够从根本上计算出来分子结构和物质的性质,这样的理论很接近于反映宇宙本质的原理,就称为第一原理了。
广义的第一原理包括两大类,以Hartree-Fork自洽场计算为基础的ab initio从头算,和密度泛函理论(DFT)计算。
也有人主张,ab initio专指从头算,而第一性原理和所谓量子化学计算特指密度泛函理论计算。
根据原子核和电子互相作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过一些近似处理后直接求解薛定谔方程的算法,习惯上称为第一原理。
新型能源材料的第一性原理计算研究

铆o Il】ateials
(2)The
aU indirect band nlaterials,and the band gaps
are
3.6eV and 3.2eV respectiVely.
gap decrease with atomic shell electronic states changing f1.0m 5d t0 4d oib砥for the
它能吸收太阳光波长范围能量,若想要充分利用太阳能中的可见光(波长在420玎rn一760nm)
部分,则带隙要足够窄,以至于可以吸收可见光光子能量。第二,半导体氧化物的价带顶要 高于O和02‘离子,而导带底要低于H和H1+离子,保证半导体光催化剂可以与水反应,使 产生的H2和02不被氧化和还原。第三,半导体光催化剂要有好的传输性。以上几个条件 是判定这种材料能否成为光催化剂材料的基本条件,要想成为良好的光催化剂材料,寻找高 转化率、高传输率和高反应率的光催化剂需要从实验和理论上进行不断的探索。 半导体氧化物InM04(M=Ta,Nb)本身具有层状结构,实验观察带隙为2.6ev和2.5ev,说明这 两种材料属于窄带隙材料,具备成为良好光催化剂的基本条件。2007年,人们又发现InNbO。
oxygen vacancy have a
displacemem,especially
for me transition metal who∞displacement signification.
Thirdly,The reSults of elec仃onic stmcture for calculation suggest
as
and their oxygen Vacancy doped.c雏es.our conclusion
第一性原理计算在材料科学中的应用

第一性原理计算在材料科学中的应用材料科学作为一门重要的学科,研究各种材料的结构和性质,为新材料的设计和开发提供理论支持。
在材料科学的研究中,第一性原理计算成为一种强有力的工具,能够帮助科学家们深入理解材料的微观结构和性质,并为材料的合成和改进提供指导。
第一性原理计算是一种基于量子力学原理的计算方法,通过求解薛定谔方程来描述材料的电子结构以及其他相关性质。
相较于传统的经验模型和半经验方法,第一性原理计算具有更高的精度和可靠性。
它不依赖于任何经验参数,完全基于物理原理,能够从头计算出材料的各种性质,如晶体结构、能带结构、电子密度分布等。
首先,第一性原理计算在材料结构预测和优化方面具有重要应用。
通过第一性原理计算,科学家们可以预测材料的晶体结构,包括晶格常数、原子位置和晶格畸变等。
这对于材料设计和合成来说具有重要意义。
例如,当科学家们希望开发新型材料或改进已有材料的性能时,他们可以通过计算不同晶体结构的能量和稳定性,找到最稳定的晶体结构,并进一步优化其性能。
其次,第一性原理计算在材料的电子结构和能带结构研究中也发挥着重要作用。
材料的电子结构决定了其物理和化学性质,如导电性、光吸收性等。
通过计算材料的能带结构和态密度,科学家们可以了解材料的电子行为和能带特征。
在探索新型半导体材料、光电材料、催化剂等领域时,第一性原理计算可提供宝贵的电子结构信息,为材料设计和性能预测提供依据。
第三,第一性原理计算还可以用于材料的物理性质预测。
材料的物理性质包括热学性质、磁学性质、光学性质等。
通过第一性原理计算,科学家们可以计算材料的声子谱、磁学性质、光学吸收谱等,进而预测材料的热传导性能、磁性和光学性能等。
这对于材料科学家来说是非常有价值的,因为他们可以通过计算预测材料的热稳定性、磁性和光学行为,并为材料的制备和应用提供方向。
最后,第一性原理计算还可以用于材料的界面和缺陷研究。
材料的界面和缺陷对其性能和功能起着重要影响。
第一性原理计算在锂硫电池中的应用进展评述

第一性原理计算是一种基于量子力学理论的计算方法,它可以用来精确计算物质的结构、性质和动态过程。
在锂硫电池中,第一性原理计算可以用来研究电池材料的电子结构、相互作用和动力学过程。
近年来,第一性原理计算在锂硫电池中的应用进展迅速。
研究人员通过第一性原理计算研究了锂硫电池材料的电子结构和相互作用,以优化电池的电化学性能。
例如,通过第一性原理计算研究锂硫电池的负极材料,可以揭示负极材料的电子结构和相互作用机制,从而优化负极材料的电化学性能。
此外,第一性原理计算还可以用来研究锂硫电池的动力学过程,如电池充放电过程中材料的相互作用和结构变化。
通过第一性原理计算研究锂硫电池的动力学过程,可以揭示电池充放电过程中材料的相互作用机制和结构变化规律,从而优化电池的电化学性能。
总之,第一性原理计算在锂硫电池中的应用进展非常快速。
近年来,研究人员通过第一性原理计算研究了锂硫电池材料的电子结构和相互作用,以优化电池的电化学性能。
此外,第一性原理计算还可以用来研究锂硫电池的动力学过程,如电池充放电过程中材料的相互作用和结构变化。
这些研究为提高锂硫电池的电化学性能和实用应用奠定了基础。
然而,第一性原理计算在锂硫电池中的应用也存在一些局限性。
例如,第一性原理计算时间复杂度较高,不能对大规模系统进行计算。
此外,第一性原理计算对系统的初始条件和边界条件要求较高,不能解决复杂的动力学过程。
因此,在应用第一性原理计算研究锂硫电池时,需要结合其它的计算方法和实验技术,以充分发挥第一性原理计算的优势。
例如,可以通过结合第一性原理计算和经典力学计算方法,来研究锂硫电池的动力学过程。
此外,可以通过结合第一性原理计算和实验技术,来验证和校正第一性原理计算的结果。
总之,第一性原理计算在锂硫电池中的应用进展迅速,为研究锂硫电池的电子结构和相互作用、动力学过程等提供了有力的手段。
但是,在应用第一性原理计算研究锂硫电池时,也应注意其局限性,并结合其它计算方法和实验技术,充分发挥其优势。
第一性原理计算方法在材料科学中的应用

第一性原理计算方法在材料科学中的应用1.引言第一性原理计算方法(First Principles Calculation)是近年来发展的新型计算方法,用于准确计算分子和固体物质的能量、结构和物理性质。
它的优势在于不依赖于实验数据,可以直接从基本原理推导出体系的特性。
在材料科学领域,第一性原理计算方法已经成为研究材料的重要工具,可以为合成新材料和设计功能材料提供理论依据,并指导实验研究。
2.第一性原理计算方法的基本原理第一性原理计算方法的基本原理是量子力学中的密度泛函理论,它的基本假设是所有粒子的运动都可以描述为波函数的运动。
根据波函数理论,一个由N个电子和原子核组成的体系的波函数可以用N个单电子波函数表示。
通过求解薛定谔方程,可以确定体系的基态能量和电子的密度,从而得到体系的性质。
3.第一性原理计算方法在材料科学中的应用(1)材料合成第一性原理计算方法可以模拟材料的结构、动力学和化学反应,为材料合成提供理论指导。
例如,使用第一性原理计算方法可以预测材料的稳定性、生长机制和晶体缺陷,从而为材料的设计和制备提供指导。
(2)材料性能第一性原理计算方法可以计算材料的电子结构、热力学性质、光电性质和磁学性质等,从而为材料的性能研究提供理论基础。
例如,通过计算材料的电子结构,可以预测材料的导电性、热导率和热电性能等,为相关应用提供指导。
(3)材料改性第一性原理计算方法可以模拟材料的界面和表面结构,研究材料的改性效果。
例如,可以通过计算材料与其他材料的界面能量来评估材料的附着性和界面稳定性,从而指导材料的改性设计。
(4)功能材料设计借助第一性原理计算方法,可以针对具体的应用需求,设计出具有特定功能的材料。
例如,通过计算材料的光电性质、催化活性和磁学性质等,可以指导材料的功能设计,为实现特定的应用提供理论指导。
4.发展趋势随着材料科学和计算科学的发展,第一性原理计算方法的应用前景越来越广阔。
未来,第一性原理计算方法将会与机器学习和高通量计算等技术结合,为材料科学的研究提供更多的可能性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
新能源材料研究中的第一性原理计算近年来,随着节能减排和环保意识的逐步加强,新能源的开发
和利用已成为世界各国共同关注的焦点。
而为了更有效地提高新
能源的利用效率和降低成本,科学家们开始转向新能源材料的研
究和开发。
在这一过程中,第一性原理计算发挥着越来越重要的
作用。
第一性原理计算是指基于量子力学理论和数学方法对材料的电
子结构和性质进行计算和模拟。
这种计算方法的好处在于既能提
供高精度的计算结果,又能对材料的微观结构和电子能带等性质
进行深入分析,为新材料的设计和开发提供有力的支持。
在新能源材料研究中,第一性原理计算可以帮助科学家们确定
材料的电子结构、晶格结构、热力学性质、光电特性等重要参数。
以太阳能电池材料为例,研究者可以通过第一性原理计算预测材
料的光吸收性能、载流子输运特性和光电转换效率等重要指标,
从而优化材料的能带结构和界面特性,提高太阳能电池的转化效率。
除了太阳能电池材料之外,第一性原理计算在其他新能源领域
的研究中也发挥着重要作用。
比如,在固态氢储存材料的研究中,
第一性原理计算可以用来预测材料的结晶形态、氢吸附能力和释放能力等关键性质,为研发更高效、更安全的氢储存材料提供支持。
在燃料电池材料的研究中,第一性原理计算可以预测氧化还原反应的能垒、电子传输特性和催化活性等参数,为提高燃料电池的效率和寿命提供重要帮助。
需要指出的是,尽管第一性原理计算具有高计算精度和深入分析的优点,但该方法也存在一些挑战和限制。
其中,计算复杂度是最主要的问题之一。
由于第一性原理计算需要对大量的原子和电子进行计算,因此计算量非常大,需要使用高性能计算机进行处理。
而由于计算复杂度高,一些材料的性质无法通过第一性原理计算来预测,需要通过实验来验证。
另一方面,第一性原理计算还需要与实验相结合,以验证计算结果的准确性和可靠性。
特别是在新能源材料研究中,第一性原理计算和实验之间的结合非常重要。
通过实验,科学家们可以验证计算结果,并不断优化计算模型,提高计算精度和可靠性。
因此,第一性原理计算和实验之间的相互补充和平衡是研究新能源材料不可或缺的环节。
综上所述,第一性原理计算在新能源材料研究中发挥着越来越重要的作用。
通过该方法,科学家们可以预测材料的电子结构和物理性质,为新材料的设计和开发提供有力的支持。
然而,由于计算复杂度高和与实验相结合的必要性,第一性原理计算的应用也面临着一些挑战和限制。
因此,科学家们需要进一步研究和优化该方法,为新能源材料的研究和开发提供更有效、更可靠的支持。