肝脏缺血再灌注损伤发生机制及防治
肝脏缺血再灌注损伤发生机制和预防

肝脏缺血再灌注损伤发生机制和预防一肝脏缺血-再灌注损伤的发生机制1反应性氧中间物的产生和释放:肝脏缺氧期间大量堆积的ATP代谢产物次黄嘌呤和氧发生反应。
产生ROI。
由于ROI在结构上即其外轨道上有一个或多个不配对电子特点,因而使ROI具有高度活性和潜在的毒性。
肝脏缺血缺氧期间产生的ROI可以通过以下几个机制损伤细胞:①选择性地损伤相邻分子如脂质、蛋白质和核酸;②增加信号传递,使嗜中性粒细胞产生趋化性和被激活;③与NO反应产生高度毒性的过氧化物[2]。
大量的动物实验和临床病例证明ROI不仅在再灌注期间明显增加,且可持续24h以上,其产生的量与再灌注损伤呈正相关。
2炎性细胞的聚集在缺血再灌注损伤的组织中有中性粒细胞在损伤区域大量聚集和黏附,其聚集和黏附能力与白三烯B4、血小板活化因子、肿瘤坏死因子、白介素1等表达有关[3-4]。
聚集和黏附的中性粒细胞可通过以下机制损伤组织器官:①聚集和黏附的中性粒细胞通过激活烟酰胺腺嘌呤二核苷酸磷酸氧化酶释放增加蛋白溶解酶降解细胞外网状结构的所有成分,破坏完整细胞和使免疫球蛋白、补体和凝血因子失去活性;②中性粒细胞释放的弹力酶与ROI可以相互作用,ROI通过蛋氨酸氧化使弹力酶的抑制剂21-抗胰蛋白酶失活;③另外ROI又可以由GMP-140等黏附分子调节后,促使粒细胞黏附于微血管内皮。
因此,肝脏缺血再灌注损伤时,粒细胞、内皮细胞黏附和ROI之间的相互作用加重了组织结构的损伤。
3血管活性物质和缺血再灌注损伤一氧化氮:一氧化氮是能够使粒细胞发生黏附作用的重要介质,是由L-精氨酸通过NO合成酶合成的,其突出的作用是松弛血管平滑肌和抑制血小板聚集[5-7]。
NO可通过以下作用机制参与肝脏的缺血再灌注损伤:①NO可与ROI竞争过氧化物歧化酶的介质,形成过氧化氮物,引起血管收缩,形成再灌注损伤中窦状隙血流停滞和无再流的现象;②NO还可通过继发性介质cGMP引起血管扩张,最终引起微循环的淤滞和再灌注损伤。
肝脏缺血-再灌注损伤的治疗进展

一
肝脏 缺血一 再灌 注 (/ 损伤 是 指缺 氧器 官 细胞 损伤 IR) 在恢复 氧供 之后更加加 重 的现象 。现将 其近 年治 疗进 展综
述 如下 。
1 IR 损 伤 发 生 机 制 /
肝脏 IR损 伤发生于外 伤 、 / 失血性 休克 、 肝切 除术 和肝
能量贮存功 能机 制 , 对持续 IR状态 下保 护肝 细胞有重要作 / 用 。缺血预处理保 护效 应 与大 鼠部分 肝脏 IR损 伤 2h内 /
肝细胞进入 细胞 循 环有 关 。预 处 理 激活 神 经生 长 因子 K B
( F K ) 应激 活化蛋 白激酶 t 8 S P p 8 和 J K 1 两者 N —B 、 o (A K 3 ) N 一, 都在循环素 D 调节上 聚合 , 细胞循环通过 G 关键点时有 D , 群 循环素促进 。N —B和 S P FK A K激活 和细 胞循 环 的生 物学 作 用 有 助 于 肝 细胞 抵 抗 缺 血 和 IR损 伤后 存 活 。Cain / l e v
学改变早 , 缺血 6 i、 0m n 再灌 注 1h时 2 %肝窦 内皮 细胞原 2
位末端标记染色 阳性 ,%肝 细胞染 色 阳性。经过 长 时间缺 2
与或介 导肝 脏损 伤。此外 , 内皮 细胞 的钙超载可导致 肝 内微
循环 阻抗增 大 , 使再灌注微循环 血液流量降低 。
血, 大部分肝 窦内皮细胞和肝细胞染色 阳性 。随着再灌 注时
促使 黄嘌呤脱氢酶 向黄 嘌呤氧化 酶转化 , 从而 为氧 自由基 的 产 生提供了催化剂 。K p e 细胞 的钙超 载是其 被激 活 的根 uf r 本原 因 , 激活 的 K p e 细胞 可通过 释放 大 量毒性 介质 而参 ufr
肝缺血再灌注损伤的发生机制及防治

8
肝胆 外科 杂志 20 0 8年 2月第 1 6卷第 1期
JunlfH p t iaySre V l 1 No Fe . 0 o ra eao l r ugr o , 6, .1, b 2 08 o bi y
,
肝 缺 血 再 灌 注 损 伤 的 发 生 机 制 及 防 治
微 循 环 阻 抗 增 大 , 再 灌 注 微 循 环 血 液 流 量 降 使 低 。
1 4 无流现 象 .
缺血 再灌 注可 引起 肝窦 内皮 细胞 的凋 亡 、 坏死 , 从 而导致 许多 肝脏 微循 环 区域血 流 的完 全停 止 。 ]
2 缺 血预 处理 与 间歇血 流 阻断
肝 缺血 后 , 脏 细 胞 供 氧 不 足 , 氧 糖 酵 解 增 肝 无 加 , 致 代 谢 性 酸 中毒 。再 灌 注 后 ,代 谢性 酸 中毒 导
迅 速 纠正 , 而 增 强 了 p 依 赖 性 酶 类 , 反 H 如磷 脂 酶 、 蛋 白水 解酶 等 的活性 , 促进 细胞 的死亡 , 种现 象称 这
有 不 可 避 免 的 血 液 丢失 。B l i 等 证 实 , 切 除 eg t hi 肝 时 , 用 间断性 肝 门 阻断 的 失 血 量是 一 次 性 肝 门 阻 采 断 的 2倍 。 缺血 预 处 理 (i h m cpeodt nn ,I s e i rcn io ig P)指 c i 预先 对组织 或 器官进 行短 暂 的 ( 5~1 n 、 引起 0mi) 不 组 织 或器 官不 可逆 损 伤 的缺 血 和 再 灌 注 , 够增 加 能
失 和 血 流 的 减 少 。 是 缺 血 再 灌 注 损 伤 的 主 要 原
因 。
其 对 随后 较 长 时 间缺 血 和 再 灌 注 损 伤 耐 受 性 的现
肝脏缺血再灌注损伤机制

3 . P MN 的活 化
、
.
经过趋化、粘 附、渗 出血管壁浸润至肝实质 内的 P MN 已经 活化。P MN 来源 的介质包括 活性氧代谢产物 ,脂类介质 ,蛋 白 酶类及各种细胞因子 。这些因子都是 P MN 的趋化、活化因子, 反过来又成为其产物,并不断地循环作用 , 加重 了肝脏和全身各 脏器的损伤 。 肝脏 的缺血 再 灌注 损伤 事 实上 就 是一 个 炎症 反应 过程 , P MN 的浸润是这一过程 中的重要组成 部分 。研 究发现缺血再 灌 注时微循环障碍的发生与中性粒细胞的激 活密切相关。 七 、细 胞 凋 亡 很 多研 究报 道肝脏缺血再灌注 中发生细胞凋亡 ,有 5 0 %~ 7 0 %的 内皮细胞和 4 0 % ̄6 0 %的肝细胞在 再灌注 中发 生凋亡 。外 膜破裂而将线粒体膜 间腔 的内容物释放 出来 , 进 而激 活一系列的 酶促级联反应导致氧化磷酸化障碍使细胞凋亡 。 八 、部分核 因子 肝缺血再灌注损伤早期枯否细胞在T N F _ a和I L - 1 B等炎性因子 作用下被激活,而这些炎性因子受 N F . K B和 A P . 1 等核转录因子的 调节。通过 T N F . Q 产生及血管内皮细胞表达 I C M - A 1 ,促使白细胞 黏附、激活导致肝再灌注损伤。N F . x B还可引起肝细胞凋亡。 九 、部分细胞 因子 主要有 I L . 1 、I L . 1 0 、T NF , a 、细 胞间粘 附分子 家族 、热休克 蛋 白、血小板激活 因子 ( P A F )等 。主要在缺血期被激活 ,参与 肝脏缺血再灌注损伤 。 综上所述 ,肝脏缺 血再灌注 损伤 是一个 多因素参与 、多种通 路共 同发挥作用 的复杂 的病理生理过程 。 通过进一步深入研究肝 脏 缺血再灌注损伤 的发生机制 , 有助于找到新的更好的预防和治 疗 H I R I 的方法 。 参考文献: [ 1 ] A r i i S ,T e r a mo t o K,K a wa mu r a C u r r e n t p r o g r e s s i n t h e
肝移植术后胆道缺血再灌注损伤的机制及防治研究进展

供体在脑死亡前 的应激状 态下 ,神经 末梢 释放大量 儿茶酚 胺 、血管紧张素 等血 管 活性 物质 ,这 些物质 使内脏血 管 收
缩 、毛 细 血 管 前 括 约 肌 痉 挛 、微 循 环 受 损 ,继 而 引 起 胆 管
细胞发生缺血性 损害 ,可见 ,胆道 缺血性 损害 在肝脏热 缺
血 之 前 已经存 在 J 。肝 移 植 手 术 过 程 中缺 血 再 灌 注 损 伤 是 不 可 避 免 的 ,尤 其 是 无 心 跳 供 体 会 使 供 肝 的 胆 道 经 历 一 个
内皮 细 胞 和 肝 细 胞 的损 伤 ,并 可 导 致 肝 脏 微 循 环 的 紊 乱 。
2 T .A P耗竭 :A P耗竭是继 发于细胞严 重缺 氧后 的病 T 理生理变化 ,可造成一系列 的细胞损 伤。包括细胞 内 C 2 a 增加 、膜磷脂 的降解 、膜 蛋 白功 能障碍 、细胞膜- 细胞 骨架
障碍 甚 至 坏 死 ’J ”。
节 ,对肝移植术 后远期 生存率 有重要 影响 ,本文 着重 在胆
道 缺 血 再灌 注 的机 制 及 防 治 进 展 作 一 综 述 。
一
、
胆 道 缺 血 再 灌 注 损 伤 的 发 生机 制
胆道缺血再灌注损伤与肝脏缺血再灌注损伤 关系密切 。
胆 道是 热 缺 血 、冷 缺 血 及 再 灌 注 等 所 致 损 伤 的重 要靶 组 织 。
细胞骨架损伤等。
缺血—再灌注损伤与缺血预处理及缺血后处理的保护作用机制(一)

缺血—再灌注损伤与缺血预处理及缺血后处理的保护作用机制(一)作者:马建伟杜会博温晓竞【关键词】缺血;再灌注损伤;缺血预处理缺血是临床上最常见的症状之一,尤其是心脏缺血损伤一直是众多学者研究和关注的问题。
既往认为短暂的心肌缺血造成的心肌可逆性损伤会使之更难以耐受再次缺血损伤。
因此认为多次短暂缺血必然发生累加而导致心肌坏死。
80年代Murry1]首次在狗的实验中发现短暂的冠脉缺血可以使心脏在经历后续长期缺血时的心梗面积较单纯长期缺血时的面积明显缩小,于是提出缺血预处理的概念。
而在2003年,Zhao等2]在犬心肌缺血后再灌注前进行了3次30s的再灌注,发现冠状动脉的内皮功能较单纯长时间再灌注得到明显改善,而且心肌梗死范围也明显缩小,其保护程度与缺血预处理相似。
因而提出了缺血后处理的概念。
这两方面的发现为缺血心肌的保护开辟了新的研究领域。
1心肌的缺血-再灌注损伤1.1心肌的缺血—再灌注损伤的概念及损伤表现缺血-再灌注(ischemiareperfusion,IR)是指心肌缺血时,心肌的代谢出现障碍,从而出现一系列功能异常;缺血一定时间的心肌再重新恢复血液供应后,心肌不一定都会恢复其正常功能和结构,反而出现心肌细胞损伤加重的表现,即所谓缺血—再灌注损伤,IRI)。
这一损伤是心脏外科、冠脉搭桥术等手术期间心肌损伤的主要因素。
其损伤表现为心肌细胞的坏死、凋亡、线粒体功能障碍、脂质过氧化物增多、自由基大量生成,并导致恶性心率失常发生,左心室收缩力减弱、室内压下降等心肌功能的抑制。
1.2心肌的缺血再灌注损伤的机制尽管几十年来人们一直在进行研究,但至今其详细的机制未被阐明,根据近年来的研究其可能的机制有:1.2.1G蛋白、腺苷酸环化酶的功能异常心肌缺血时,对于G蛋白、腺苷酸环化酶活性的变化各家报道不一,有研究表明在体大鼠缺血区G蛋白含量明显降低3],有结果表明,离体大鼠缺血区G蛋白含量无明显变化4],也有结果表明,在体狗心肌缺血时,心肌G蛋白含量出现明显增加5]。
缺血再灌注损伤机制

缺血再灌注损伤机制缺血再灌注损伤(ischemia-reperfusion injury)是一种普遍存在的生理现象,常见于心血管外科手术、心肌梗死、脑中风等各种临床情况中。
本文将以缺血再灌注损伤机制为主题,从深度和广度两个方面探讨该主题的各个方面,以帮助读者更全面地理解这一现象。
一、缺血再灌注损伤的基本概念缺血再灌注损伤指的是当组织或器官遭受缺血(血液供应中断)一段时间后,再次供血恢复时所引发的损伤反应。
尽管再灌注的目的是恢复局部供血,但却可能对组织或器官造成更严重的伤害,导致细胞坏死、炎症反应和功能丧失等不良后果。
二、缺血再灌注损伤的机制1. 氧化应激和自由基产生在缺血时,组织或器官缺乏氧气和能量供应,导致线粒体功能障碍和ATP合成降低。
当再灌注发生时,由于血液中大量的氧气重新供应,导致活化的线粒体释放更多反应性氧种和自由基,从而引发氧化应激反应,破坏细胞膜和细胞器功能。
2. 炎症反应激活缺血再灌注损伤可引发炎症反应,释放细胞因子、趋化因子和炎症介质,进一步导致炎症细胞浸润、血管扩张和血小板聚集等炎症反应。
这些炎症反应激活了免疫细胞和炎性细胞,进一步加剧了组织损伤。
3. 钙离子紊乱缺血再灌注损伤会导致细胞内和细胞外钙离子浓度失衡,破坏细胞内钙离子平衡和细胞外钙离子浓度梯度。
这种钙离子紊乱会引发线粒体功能失调、细胞凋亡和细胞死亡等多种病理生理过程。
4. 血管内皮功能损伤缺血再灌注损伤可导致血管内皮细胞的受损和功能异常,进而引发血管扩张、血小板聚集和血管渗透性增加等现象。
这些改变会进一步造成血管内皮功能的破坏,加重缺血再灌注损伤。
三、缺血再灌注损伤防治策略1. 保护组织氧供在缺血再灌注过程中,保持良好的氧供对减轻损伤非常重要。
提前做好血液输注、氧气供应和改善心血管循环等措施,可以有效预防缺血再灌注损伤的发生。
2. 抗氧化治疗应用抗氧化剂,如维生素C、维生素E和谷胱甘肽等,可以减轻缺血再灌注引起的氧化应激反应。
缺血再灌注损伤机制
PARALLEL研究是迄今为至规模最大的曲美他嗪随机对照研究。 PARALLEL研究是迄今为至规模最大的曲美他嗪随机对照研究。该研究入 研究是迄今为至规模最大的曲美他嗪随机对照研究 选903例稳定性心绞痛患者,在β受体阻滞剂的基础上随机加用曲美他 903例稳定性心绞痛患者, 例稳定性心绞痛患者 嗪或长效硝酸酯(硝酸异山梨酯)治疗12周 嗪或长效硝酸酯(硝酸异山梨酯)治疗12周,结果联合曲美他嗪组每周 12 心绞痛次数较基线时显著降低75.9%, 心绞痛次数较基线时显著降低75.9%,每周硝酸甘油用量较基线时显著 75.9% 降低78.8%,且显著低于联用硝酸酯组。 降低78.8%,且显著低于联用硝酸酯组。 78.8%
结
果
1.显著降低心绞痛发作次数(75.9% 61.6%,p<0.0001) %,p<0.0001 1.显著降低心绞痛发作次数(75.9%比61.6%,p<0.0001) 2.减少硝酸甘油用量(78.8% 63.2%,p<0.0001) %,p<0.0001 2.减少硝酸甘油用量(78.8%比63.2%,p<0.0001) 3.联用曲美他嗪抗心绞痛作用更强,尤其对60岁以 3.联用曲美他嗪抗心绞痛作用更强,尤其对60岁以 联用曲美他嗪抗心绞痛作用更强 60 上的患者及糖尿病患者 4.曲美他嗪组对稳定型心绞痛患者的生活质量的 4.曲美他嗪组对稳定型心绞痛患者的生活质量的 改善更佳 5.曲美他嗪组不良反应发生率显著降低。 5.曲美他嗪组不良反应发生率显著降低。 曲美他嗪组不良反应发生率显著降低
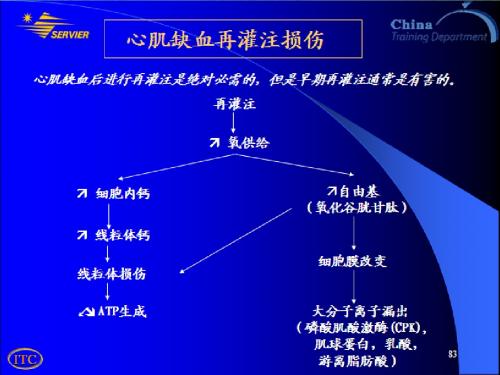
因缺血、 生成减少, 因缺血、缺氧使 ATP 生成减少,钙离子进入细胞 增多,使细胞膜损伤,以及线粒体功能受损。 增多,使细胞膜损伤,以及线粒体功能受损。 1.再灌注时,氧气的增多,就会生成大量的氧自由 1.再灌注时,氧气的增多,就会生成大量的氧自由 再灌注时 基。由于线粒体功能此时尚未恢复,所以对于氧自由基 由于线粒体功能此时尚未恢复, 的 清除能力不足,导致氧自由基增多。氧自由基的增 清除能力不足,导致氧自由基增多。 多,就会对膜磷脂造成损伤,包括细胞膜,细胞器膜 就会对膜磷脂造成损伤,包括细胞膜, 如线粒体膜,溶酶体膜,内质网膜等等的损伤, 如线粒体膜,溶酶体膜,内质网膜等等的损伤,会进 一步损伤细胞。同时氧自由基的增多还会对蛋白质, 一步损伤细胞。同时氧自由基的增多还会对蛋白质, 核酸及细胞外基质造成损伤,从而加重了细胞的凋亡 加重了细胞的凋亡。 核酸及细胞外基质造成损伤,从而加重了细胞通透性增强, 2.再灌注时,由于细胞膜的损伤,通透性增强,使大量的 再灌注时 钙离子顺浓度梯度内流,造成细胞内钙超载 钙超载; 钙离子顺浓度梯度内流,造成细胞内钙超载;同时线 粒体功能的障碍,ATP生成减少, 粒体功能的障碍,ATP生成减少,肌膜及肌浆网膜钙泵 生成减少 功能障碍,不能排出和摄取细胞浆中过多的钙,致使 功能障碍,不能排出和摄取细胞浆中过多的钙, 细胞浆中游离钙浓度增加而进一步造成钙超载。 细胞浆中游离钙浓度增加而进一步造成钙超载。而钙 超载会进一步加重细胞的凋亡。 超载会进一步加重细胞的凋亡。 加重细胞的凋亡
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
缺血-再灌注时的钙超载发生机制
①Na+/Ca2+交换反向转运增强。缺血引起 的细胞内高Na+、高H+、PKC激活可直接或 间接激活Na+/ Ca2+交换蛋白反向转运,将 大量Ca2+运入胞浆; ②生物膜损伤:细胞膜、线粒体及肌浆网 膜损伤,可使钙内流增加和向肌浆网转运 减少。
①微血管内血液流变学改变 ②微血管管腔狭窄,阻碍血液灌流; ③微血管通透性增高 ④激活的中性粒细胞与血管内皮细胞可释放
致炎物质,损伤组织细胞。
3 氧自由基和肝脏缺血再灌注损伤
再灌注期缺血组织恢复血氧供应的同时也提 供了大量电子受体,使氧自由基在短时间内爆发 性增多。主要途径有:
①内皮细胞源。经黄嘌呤氧化酶催化嘌呤类代谢并 释放出大量电子,为分子氧接受后产生活性氧;
②中性粒细胞源。再灌注期激活的中性粒细胞产生 大量氧自由基,称为呼吸爆发;
③线粒体源。线粒体氧化磷酸化功能障碍,进入细 胞内的氧经单电子还原而形成的氧自由基增多, 而经4价还原生成的水减少。
3 氧自由基和肝脏缺血再灌注损伤
自由基具有极活泼的反应性,一旦生成可 经其中间代谢产物不断扩展生成新的自由 基,形成连锁反应。 自由基可与磷脂膜、蛋白质、核酸和糖类 物质反应,造成细胞功能代谢障碍和结构 破坏。
肝脏缺血再灌注损伤的发生机制 及防治进展
中山大学附属三院麻醉科
肝缺血再灌注损伤( IRI) 是肝脏 外科中常见的病理过程,多见于需 要肝断流的肝脏外科手术,如失血 性休克、肝叶切除、肝移植等
我科的研究方向-肠、脑、肝缺血。
一 肝脏缺血再灌注损伤的发生机制
1 钙超载 2 中性粒细胞 3 氧自由基
钙超载和肝脏缺血再灌注损伤
细胞因子
国内研究在鼠肝脏部分缺血再灌注损伤过程 中预先给予参附,通过增加抑炎因子IL-10水 平,降低TNF-α,下调其介导的固有免疫应 答最终减轻肝脏缺血再灌注损伤。 Bibo Ke在肝脏部分缺血再灌注过程中通过 IL-10基因转染小鼠使小鼠血清IL-10 水平增 高导致肝细胞损伤减轻 。
Bibo Ke,Xiu-Da Shen,Sei-Ichiro Tsuchihashi,et al. Human Gene Therapy,2007,18 ( 4 ):355-366.
血红素氧合酶系统
CO 在缺血再灌注损伤中的细胞保护作用其 他的机制可能还有:抑制诱生型一氧化氮 合酶(iNOS),抑制内皮细胞凋亡。 辅以HO-1 竞争性抑制物锌原卟啉发现外源 性CO 能完全取代内源性HO-1 抑制肝脏缺 血再灌注损伤。因此,HO-1 介导的细胞保 护抗缺血再灌注损伤需要外源性CO 的产生, 并且完全能够被CO 替代。
肝脏缺血再灌注损伤的发生机制新进展
Kupffer细胞激活 细胞因子 细胞级联反应 T淋巴细胞 肝血红素氧合酶系统(HO-1)
Kupffer细胞激活—细胞因子的释放
Kupffer 细胞是一种与肝脏缺血再灌注损伤 有关的非实质细胞, 这种驻留型巨噬细胞 可出现在肝脏的窦状隙内,肝Kupffer 细胞 在肝脏再灌注时被激活, 产生一系列炎症 性细胞因子(PGs,PAF,IL-1,TNF-α, IL-6,IFN-γ)和氧自由基,这些作为直接 的细胞毒素作用于内皮细胞和肝细胞,导 致肝脏损伤。
越来越多的证据表明T 细胞在介导缺血再灌注引起的短 期和长期损伤中起重要作用,全身性免疫抑制剂可减轻肝细 胞缺血再灌注后损伤,提示T 淋巴细胞参与了损伤的病理生 理过程。 Shen 等发现T 淋巴细胞缺陷小鼠肝脏缺血再灌注后损伤较 对照组明显减轻。 Khandoga 等发现在再灌注早期淋巴细胞在肝窦状隙发生附 着并随着冷缺血时间的延长损伤肝脏功能。 Moine等发现抗炎症介质IL-10 在缺血再灌注损伤过程中具 有保护作用,不但与抑制Kupffer 细胞释放细胞因子有关, 而且与抑制T 细胞驻留有关。 所有这些研究结果与T 细胞在缺血再灌注损伤机制中起主导 作用相符合
细胞级联反应
肝脏缺血再灌注损伤的过程是许多相关因 子联合作用产生级联反应导致最后的肝脏 功能衰竭。 大量的证据证明Kupffer 细胞、中性白细胞、 内皮细胞以及反应性氧族在缺血再灌注损 伤的发病机制中起了重要作用,这种相关 的级联反应过程导致最终结果是组织结构 的改变并引起肝细胞功能的衰竭
T 淋巴细胞
血红素氧合酶系统
血红素氧合酶是普遍存在的酶,在亚铁血 红素分解为胆绿素、一氧化碳和自由铁的 过程中起催化作用。 HO 系统的激活在缺血再灌注触发的级联反 应中可能提供了细胞保护功能。 近年来的发现认为HO 途径不仅抗氧化,而 且是作为更加复杂的细胞保护和免疫调节 系统。
Fondevila C, Shen XD et al. Biliverdin therapy protects rat livers from ischemia and reperfusion injury[J].Hepatology,2004,40(6):1333-1341.
血红素氧合酶系统
由亚铁血红素释放的CO 可能在不同的细胞 和生物学过程中作为一种调控分子,类似 于NO。这两种物质引起平滑肌松弛,导致 内皮组织血管舒张,这可能是CO 介导的抗 缺血再灌注损伤的细胞保护作用的机制。 在灌注时CO 通过抑制血管收缩维持微循环 血流,减少缺血再灌注损伤相关的微血管 血栓形成。
钙超载引起再灌注损伤机制
①线粒体功
②激活酶类。 Ca2+浓度升高可激活磷脂酶、 蛋白酶、核酶等,促进细胞损伤;
③促进氧自由基生成; ④再灌注性心律失常; ⑤肌原纤维过度收缩。
2 中性粒细胞和肝脏缺血再灌注损伤
激活的中性粒细胞与血管内皮细胞相互作用, 造成微血管和细胞损伤。
Oltean M, Pullerits R, Zhu C, et al. Donor pretreatment with FK506 reduces reperfusion injury and accelerates intestinal graft recovery in rats[J]. Surgery,2007,141(5):667-677.