缬沙坦常用杂质中国药典、EP和USP名称对比
CP、USP炽灼残渣和EP硫酸灰分的区别

EP2.4.14硫酸灰分硫酸灰分:在600±50℃下炽灼坩埚30min,在放有硅胶的干燥器中冷却并称重。
取规定量的供试品,置于坩埚(如供试品分子中含有碱金属或氟元素,则应使用铂坩埚)中并称重。
用少量硫酸(通常1ml)润湿供试品,然后在低温下缓慢加热直至样品全部炭化。
冷却后,用少量硫酸(通常1ml)润湿残渣,在低温加热直至白烟不再产生,然后在600±50℃下炽灼直至残渣完全成灰。
要保证火焰在整个实验过程的任何阶段都不产生。
把坩埚置于成有硅胶的干燥器中冷却,再次称重并计算残渣的百分含量。
如果得到的残渣的量超过规定限度,用硫酸重复润湿并灼烧,每30min作为一个阶段直至连续两次称重的差不超过0.5mg或者残渣的百分含量符合规定限度。
所选择的待测物质的量(通常1-2g)为的是残渣的质量(通常约1mg)在规定限度下能够被测定并具有充分的准确度。
USP<281>炽灼残渣方法:在600±50℃下炽灼坩埚30min,在放有硅胶的干燥器中冷却并准确称重。
精确称取1-2g供试品,置于坩埚内。
用少量硫酸(通常1ml)润湿供试品,然后在低温缓慢加热直至样品全部炭化。
冷却;用少量硫酸(通常1ml)润湿残渣,在低温加热直至白烟不再产生,然后在600±50℃下炽灼直至残渣完全成灰。
要保证火焰在整个实验过程的任何阶段都不产生。
把坩埚置于成有硅胶的干燥器中冷却,准确称重并计算残渣的百分含量。
除非另有规定,如果得到的残渣的量超过规定限度,用硫酸重复润湿并灼烧,每30min作为一个阶段直至连续两次称重的差不超过0.5mg或者残渣的百分含量符合规定限度。
操作灼烧时要在通风橱下进行,但要避免气流影响炭的灼烧。
也可以用马弗炉,首次在600±50℃下的炽灼用它最好。
CP 附录ⅧN 炽灼残渣检查法取供试品1.0-2.0g或各品种项下规定的重量,置已炽灼至恒重的坩埚(如供试品分子中含有碱金属或氟元素,则应使用铂坩埚)中,精密称定,缓缓炽灼至完全炭化,放冷;除另有规定外,加硫酸0.5-1ml是湿润,低温加热至硫酸蒸汽除尽后,在700-800℃炽灼使完全灰化,移至干燥器内,放冷,精密称定后,再在700-800℃炽灼至恒重,即得。
USP方法与EP方法对比实验

USP与EP检测方法的对比实验一实验目的:比较USP与EP检测方法结果的区别二实验中需要注意的重要参数:仪器:高效液相色谱仪,紫外分光光度计,TLC,傅里叶红外变换光谱仪,*同种仪器使用同一台试剂:同种规格的试剂应尽量使用同一生产批号或同一批配制的。
人员:同一指标必须由同一人完成。
三执行方法及标准:1 定性鉴别:1.1 EP方法:A红外吸收光谱在105o C下干燥样品6小时,用4mg样品录制谱图。
将所得谱图与化学参比物谱图。
B:在0.4ml溶液S1中加入10ml水,5ml稀盐酸,2ml重铬酸钾溶液。
产生橘黄色沉淀。
C:在1ml的S1溶液中加入0.2ml的二甲氨基苯甲醛和0.1ml的硫酸,生成粉红色溶液。
D:在0.1ml的S1溶液中加入5ml的水和0.2ml0.05mol/l的碘溶液,生成红色溶液。
E:取0.5g 样品溶解于10ml水中,摇动,全部样品溶解。
溶液外观:溶液S澄清,颜色浅于B6,BY6。
1.2 USP方法:A.取10ml20mg/ ml的样品溶液加入20ml1当量的盐酸和5ml的重铬酸钾溶液,生成黄色沉淀。
B.用2ml纯水溶解75mg的硝酸钴和300mg的硫氰酸铵,加入5ml的20mg / ml 的样品溶液,再加入3当量的盐酸,生成蓝色沉淀。
C.取5 ml 5mg/ ml的样品溶液加入数滴碘液,溶液变成深红色。
注:溶液S:取1.0g样品,溶解于无二氧化碳水中并稀释至20ml。
取少量待测样品到水中,用磁力搅拌器搅拌。
溶液S1:取2.5g样品,溶解于无二氧化碳水,并稀释至25ml。
取少量待测样品到水中,用磁力搅拌器搅拌。
参比液B6溶液的配:黄色溶液:用盐酸溶液(HCl/H2O=25ml/975ml)溶解46g FeCl3并定容至1000ml,滴加相同的盐酸溶液调整FeCl3·6H2O的浓度为45.0 mg/ml,避光存放。
滴定:加10.0ml 上述溶液,15ml水,5ml HCl,4gKI于250ml带塞磨口锥形烧瓶中,塞上瓶塞,加100ml水,避光存放15min。
HPLC法测定缬沙坦胶囊的有关物质

HPLC法测定缬沙坦胶囊的有关物质发表时间:2013-02-25T17:15:14.263Z 来源:《医药前沿》2012年第36期供稿作者:李明杰蒋燕杰郭中明罗兆亮[导读] 该方法准确可靠,专属性强,可用于缬沙坦胶囊有关物质测定。
李明杰蒋燕杰郭中明罗兆亮(山东罗欣药业股份有限公司 276000)【摘要】目的建立测定缬沙坦胶囊有关物质的HPLC方法。
方法用Diamonsil C18(250mm×4.6mm,5μm)色谱柱,以乙腈-水-冰醋酸(500:500:1)为流动相,流速1.0ml?min~(-1),柱温40℃,检测波长230nm,进样量20μl。
结果有关物质各杂质峰与主峰之间的分离良好,且已知单个杂质分离良好。
结论该方法准确可靠,专属性强,可用于缬沙坦胶囊有关物质测定。
【关键词】缬沙坦胶囊有关物质高效液相色谱仪缬沙坦是一种口服有效的特异性的血管紧张素Ⅱ(AT1)受体提起拮抗剂,它选择性地作用于AT1受体亚型,阻断AngⅡ与AT1受体的结合(其特异性拮抗AT1受体的作用大于AT2受体约20,000倍),从而抑制血管收缩和醛固酮的释放,产生降压作用。
临床用于治疗轻、中度原发性高血压[1]。
《中国药典》2010年版收载了缬沙坦原料药及胶囊的含量和有关物质测定方法,本次实验采用《中国药典》2010年版HPLC法对缬沙坦胶囊进行有关物质方法学研究,并与国内上市缬沙坦胶囊质量作了系统的比较。
1 方法与结果1.1 色谱条件与系统适用性[2]色谱柱:Diamonsil C18(250mm×4.6mm,5μm);柱温:30°C;流动相:乙腈-水-冰醋酸(500:500:1);流速:1.0ml/min;进样量:20μl;检测波长:230nm。
1.2 专属性试验空白干扰试验:精密量取空白溶剂20μl,注入液相色谱仪,记录色谱图。
结果表明,空白溶剂不干扰本品的有关物质测定。
按处方配制不含缬沙坦的空白样品,作为供试品溶液。
USP方法与EP方法对比实验

USP方法与EP方法对比实验USP方法与EP方法是药物品质控制中常用的两种方法,主要用于评估药品的纯度和质量稳定性。
两种方法在原理、应用范围、操作步骤和结果解释等方面存在一些差别。
本文将对USP方法和EP方法进行比较,并分析它们的优点和缺点。
一、原理和应用范围二、操作步骤三、结果解释在结果解释方面,两种方法也有所不同。
USP方法通常使用定性分析,即将待测物与参比品进行对比,根据是否符合一定标准来评估药品的质量。
而EP方法更注重定量分析,通过测量待测物与参比品之间的定量差别来评估药品的质量。
四、优点和缺点P方法的优点包括:(1)简单易行:USP方法通常使用常规分析方法,如比色反应或重量法,操作相对简单易行,适用于许多药品的质量评估。
(2)成熟可靠:USP方法由美国药典委员会制定,经过了长期的实践检验,成熟可靠,被广泛使用于全球各个药品生产和监管领域。
(3)范围广泛:USP方法适用于多种药物的分析和检测,无论是固体药品、液体制剂还是注射剂,都可以使用USP方法进行质量控制。
P方法的缺点包括:(1)定性分析:USP方法主要是定性分析,无法提供对待测物的定量信息。
这对于一些要求精确测量的药品来说,可能不够准确。
(2)结果解释灵活性较低:USP方法通常需要根据一定的标准来判断样品的合格与否,标准的制定较为严格,导致结果解释的灵活性较低。
(3)更新滞后:USP方法更新较慢,这意味着在一些新药和新技术的出现之前,USP方法可能无法及时应用于对这类药品的质量控制。
3.EP方法的优点包括:(1)定量准确:EP方法以定量分析为主,通过测量待测物与参比品的定量差别来评估待测物的质量,提供了更准确的定量信息。
(2)灵活可变:EP方法的结果解释灵活性较高,可以根据实际情况进行灵活调整和解释。
这对于研发新的药物和新的质量控制方法具有一定的优势。
(3)更新快速:EP方法更加注重创新和发展,能够及时采用新的分析方法和技术,以适应不断更新的药品质量控制要求。
缬沙坦质量标准
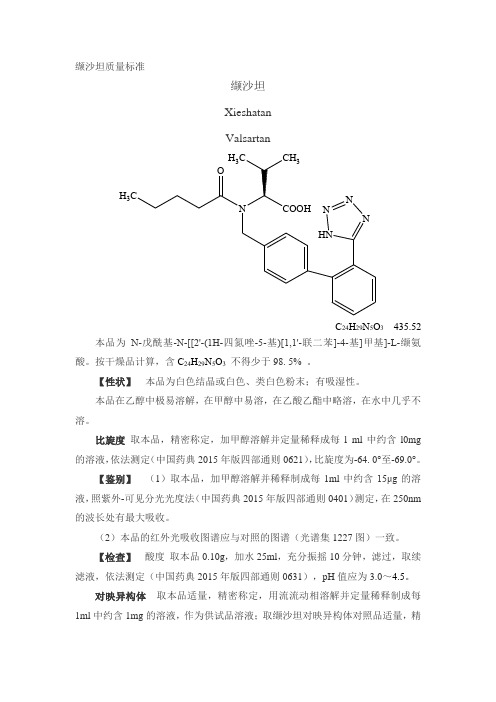
缬沙坦质量标准缬沙坦 XieshatanValsartanC 24H 29N 5O 3 435.52本品为N -戊酰基-N -[[2'-(1H -四氮唑-5-基)[1,1'-联二苯]-4-基]甲基]-L -缬氨酸。
按干燥品计算,含C 24H 29N 5O 3 不得少于98. 5% 。
【性状】 本品为白色结晶或白色、类白色粉末;有吸湿性。
本品在乙醇中极易溶解,在甲醇中易溶,在乙酸乙酯中略溶,在水中几乎不溶。
比旋度 取本品,精密称定,加甲醇溶解并定量稀释成每l ml 中约含l0mg 的溶液,依法测定(中国药典2015年版四部通则0621),比旋度为-64. 0°至-69.0°。
【鉴别】 (1)取本品,加甲醇溶解并稀释制成每1ml 中约含15μg 的溶液,照紫外-可见分光光度法(中国药典2015年版四部通则0401)测定,在250nm 的波长处有最大吸收。
(2)本品的红外光吸收图谱应与对照的图谱(光谱集1227图)一致。
【检查】 酸度 取本品0.10g ,加水25ml ,充分振摇10分钟,滤过,取续滤液,依法测定(中国药典2015年版四部通则0631),pH 值应为3.0~4.5。
对映异构体 取本品适量,精密称定,用流流动相溶解并定量稀释制成每1ml 中约含1mg 的溶液,作为供试品溶液;取缬沙坦对映异构体对照品适量,精H 3H 3CO密称定,用流动相溶解并定量稀释制成每1ml中约含0.01mg的溶液,作为对照品溶液;另分别取缬沙坦对照品和缬沙坦对映异构体对照品适量,精密称定,用流动相溶解并定量稀释制成每1ml中约含0.04mg缬沙坦和0.04mg缬沙坦对映异构体的溶液,作为系统适用性溶液。
照高效液相色谱法(中国药典2015年版四部通则0512)测定,用手性色谱柱(Silica gel OD for chiral separation R),如Chiralcel Daicel OD-H,4.6×250mm或相当规格;以正己烷-异丙醇-三氟醋酸(850:150:1)作为流动相,检测波长为230nm,流速为0.8m/min。
中国药典和美国药典中的细菌内毒素检测法的不同点比较

灵敏度复核实验
未说明
至少用每批鲎试剂中的1支试剂进行灵敏度复核实验
当最大浓度2.0λ管为阳性,最低浓度0.25λ管均为阴性,阴性对照管为阴性时,验方为有效
浓度最低的标准品溶液的所有重复管均为阴性,实验方为有效
按下式计算反应终点浓度的几何平均值,即为鲎试剂灵敏度的测定值(λc):
λc=lg-1(ΣX/4)
供试品溶液的制备
某些供试品需进行复溶、稀释或在水性溶液中浸提
用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提
对于过酸、过碱或本身有缓冲能力的供试品需调节被测溶液(或其稀释液)的pH值,一般要求供试品溶液的pH值在6.0-8.0的范围内
如果需要,可调节待测溶液(或其稀释液)的pH值,以使鲎试剂和样品的混合物的pH范围落在鲎试剂生产商指定的范围内,这通常适用于pH值在6.0-8.0范围内的产品
与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水
用于细菌内毒素定量测定用的细菌内毒素检查用水,内毒素含量应小于0.005EU/ml
未提到
实验用具的准备
实验所用器皿需经处理,除去可能存在的外源性内毒素,常用的方法是250℃下干烤至少1小时,也可用其它确证不干扰细菌内毒素检查的适宜方法。
使用经验证的除热原程序对所有玻璃器皿和遇热稳定的材料在热空气烘箱中进行除热原,常用的最低温度和最少的时间是250℃下30分钟
中国药典和美国药典中的细菌内毒素检测法的不同点比较requirementscpusp细菌内毒素工作标准品cse对cse的用途及效价进行定义细菌内毒素工作标准品中每1ng细菌内毒素的效价应不小于2eu不大于50eu未提到cse细菌内毒素检查用水与灵敏度为003euml或更高灵敏度的鲎试剂在371条件下24小时不产生凝集反应的灭菌注射用水与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水用于细菌内毒素定量测定用的细菌内毒素检查用水内毒素含量应小于0005euml未提到实验用具的准备实验所用器皿需经处理除去可能存在的外源性内毒素常用的方法是250下干烤至少1小时也可用其它确证不干扰细菌内毒素检查的适宜方使用经验证的除热原程序对所有玻璃器皿和遇热稳定的材料在热空气烘箱中进行除热原常用的最低温度和最少的时间是250下30分钟试验操作过程应防止微生物污染未提到内毒素标准品贮液的制备未提到usp的rse的效价定为10000uspeu支用5ml鲎试剂检查用水复溶部内容物用漩涡混合器间歇混合30分钟并用此原液作系列稀释将原液置于冰柜中保存不超过14天作以后的稀释之用在使用前用漩涡混合器强力混合不少于钟在作下一步稀释前需对前面的稀释液混合不少于30秒不要贮存稀释液因为没有数据能证明其不会因为吸附作用而失去活性
各国药典溶解性EP USP ChP差异比较
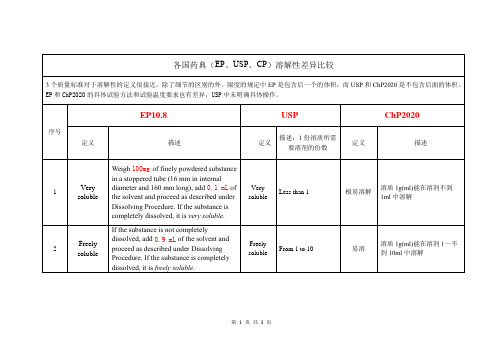
From 10 to 30
溶解
溶质1g(ml)能在溶剂10~不到30ml中溶解
4
Sparingly soluble
If the substance is not completely dissolved, add7.0 mLof the solvent and proceed as described under Dissolving Procedure. If the substance is completely dissolved, it issparingly soluble.
Very soluble
Less than 1
极易溶解
溶质1g(ml)能在溶剂不到1ml中溶解
2
Freely soluble
If the substance is not completely dissolved, add0.9 mLof the solvent and proceed as described under Dissolving Procedure. If the substance is completely dissolved, it isfreely soluble.
Practically insoluble, or insoluble
10,000 and over
几乎不溶或不溶
溶质1g(ml)在溶剂10000ml中不能完全y for 1 min and place in a constant temperature device, maintained at a temperature of 25.0 ± 0.5 °C for 15 min. If the substance is not completely dissolved, repeat the shaking for 1 min and place the tube in the constant temperature device for 15 min.
缬沙坦胶囊处方工艺筛选及溶出度评价

缬沙坦胶囊处方工艺筛选及溶出度评价摘要:缬沙坦胶囊属于血管紧张素ⅱ受体拮抗剂的抗高血压药,用于治疗轻、中度原发性高血压。
通过对缬沙坦胶囊处方中交联聚维酮、聚维酮k30和十二烷基硫酸钠的含量进行调节,比较自制制剂(处方1-3)和原研制剂(批号:x1282)的f2因子,得到当处方中交联聚维酮、聚维酮k30和十二烷基硫酸钠含量分别为9.81%、9.65%、0.65%时和原研制剂比较相似。
关键词:缬沙坦处方工艺溶出度评价中图分类号:r927 文献标识码:a 文章编号:1007-3973(2013)003-075-021 仪器、试剂和试药溶出仪:rcz-8型药物溶出度仪(上海黄海药检仪器有限公司);紫外可见分光光度计:tu-1810紫外可见分光光度计(北京谱析通用仪器有限责任公司)。
水:娃哈哈,纯净水;磷酸二氢钾:天津市恒兴化学试剂制造有限公司,分析纯;氢氧化钠(片状):天津市巴斯夫化工有限公司,分析纯;三水醋酸钠:天津市福晨化学试剂厂,分析纯;冰醋酸:天津市富于精细化工有限公司;盐酸:北京化工厂,分析纯。
缬沙坦对照品:中国生物制品检定所,批号:100651-201102;原研缬沙坦胶囊:北京诺华制药有限公司,80mg,批号:x1282 ;微晶纤维素、交联聚维酮、聚维酮k30、十二烷基硫酸钠、硬脂酸镁:上海昌为医药辅料技术有限公司。
2 制备工艺筛选2.1 处方量根据国外原研制剂专利,确定规格和辅料种类,设计处方见表1,分别制1000粒。
表1 处方筛选1000粒处方量(g)2.2 工艺过程(1)取适量缬沙坦和各辅料过80目筛;(2)称取已过80目筛的聚维酮k30、十二烷基硫酸钠,加温水溶解至50ml,作为粘合剂;(3)称取已过80目筛的缬沙坦、微晶纤维素101、交联聚维酮,以等量递增法混合均匀,加粘合剂制软材,过18目筛制粒,在80℃下干燥2h,用20目筛整粒,再加入硬脂酸镁,混合均匀;(4)用3号胶囊填充。