药物设计学完整版
药物设计学完整版

药物设计学完整版药物设计学是一门研究如何设计出具有特定生物活性的化合物的学科,它结合了化学、生物学、计算机科学等多个领域的知识。
药物设计的目标是通过理解疾病发生发展的分子机制,找到能够干扰或调节这些过程的化合物,从而开发出新的治疗方法。
药物设计学的主要内容包括:1. 目标选择:选择合适的药物靶点,即与疾病相关的生物分子,如蛋白质、核酸等。
2. 先导化合物的发现:通过高通量筛选、计算机辅助设计等方法,寻找具有潜在生物活性的化合物。
3. 优化和改造:对先导化合物进行结构改造,以提高其生物活性、选择性和药代动力学性质。
4. 体外和体内活性测试:评估化合物的生物活性、毒性和药代动力学性质。
5. 临床前研究:对候选药物进行安全性、有效性和药代动力学研究。
6. 临床试验:在人体中评估候选药物的安全性和有效性。
7. 上市后监测:对上市药物进行长期的安全性、有效性和经济性监测。
药物设计学的发展历程可以追溯到20世纪初,当时人们开始尝试通过化学方法合成具有生物活性的化合物。
随着生物学、计算机科学等领域的不断发展,药物设计学逐渐形成了一门独立的学科。
近年来,随着高通量筛选、计算机辅助设计等技术的进步,药物设计学的发展速度大大加快,越来越多的新药被成功开发出来。
药物设计学的研究成果对于新药研发具有重要意义。
通过药物设计学的研究,我们可以更加深入地理解疾病发生发展的分子机制,找到更加有效的治疗方法。
同时,药物设计学的研究还可以推动化学、生物学、计算机科学等领域的交叉融合,促进这些领域的发展。
药物设计学完整版药物设计学是一门研究如何设计出具有特定生物活性的化合物的学科,它结合了化学、生物学、计算机科学等多个领域的知识。
药物设计的目标是通过理解疾病发生发展的分子机制,找到能够干扰或调节这些过程的化合物,从而开发出新的治疗方法。
药物设计学的主要内容包括:1. 目标选择:选择合适的药物靶点,即与疾病相关的生物分子,如蛋白质、核酸等。
药物设计学-导论
配置
提取 新药创新 运用 知识 规则 转化 模拟 信息
化学信息学在新药创新中的运用
图式如下:
从大量的数据中提取对发展药物有用的规则
转换
知识 信息**
规则 事实
提取 (1)文献资料* 数 (2)制药、化学品和数据库公司积累 据 (3)组合化学库和高通量筛选
数 目
*媒体形式的表现、记录和管理( CA) **化学物质的理化性质、反应性(具有实践意义)
△(三)21世纪
[1] ME Wolff ed. Berger’s Medicinal Chemistry and Drug Discovery, 6th Ed, 2003, VolⅠ: Drug Discovery, and VolⅡ: Drug Discovery and Drug Development. [2] HJ Smith ed. Smith and Williams' Introduction to the Principles of Drug Design and Action,4th Ed, 2004. [3] RB Silverman ed. The Organic Chemistry of Drug Design and Drug Action,2004. [4] D Triggle and J Taylor ed. Comprehensive Medicinal ChemistryⅡ, Vol 1,2,3,4,5,8, 2006~2007. [5] 张礼和主编导读版,“药物化学百科丛书”第1,2,3,4,5,6,7,8,9,10, 11,16册,科学出版社,2007 [6] 迟玉明主译. 创新药物化学,世界图书出版公司,2005. [7] 仇缀百主编. 药物设计学(第二版),高等教育出版社,2008.
药物设计学

药物设计学药物设计学是一门涉及化学、生物学、医学等学科知识的学科,其核心是通过理性设计化合物的结构,来达到治疗疾病的目的。
药物设计学包括从已知活性分子出发,结合分子的构效关系和药物代谢动力学、毒理学等方面的知识,设计具有更佳活性,更佳生物利用度和更佳安全性的新化合物,以满足临床治疗的需要。
一、药物研发的阶段药物设计学贯穿于一系列关键的药物研发阶段,如药物发现、药物优化、药物制备、药物评价等阶段。
其中,药物发现阶段可以进一步划分为高通量筛选、药物分子设计和计算机辅助药物设计等子阶段。
药物优化阶段,则是通过对药物分子进行结构优化、化学修饰等方式,以优化药物的活性、药代动力学和毒理学性质,并选择最适宜的给药途径,提高药物的疗效和安全性。
药物制备阶段,目的是制造有效、可重复生产的药物成品,并保证其品质符合药理学、毒理学和药代动力学特性的要求。
药物评价阶段,则涉及各类体外和体内试验、临床实验等,以验证药物的有效性、安全性、药代动力学等药物特性。
二、药物分子设计方法药物分子设计方法是药物设计学的核心之一。
主要分为定量构效关系(QSAR)、配体基本位点亲和力模型和分子基本位点亲和力模型等方法。
定量构效关系(QSAR)的方法是在一定的条件下,通过计算一系列分子性质的参数,构建参数与活性(或毒性)之间的定量关系模型,并进行预测。
配体基本位点模型则是从药物分子中提取出与生物靶分子相互作用的关键位点信息,以提高理性设计药物分子的精准性。
分子基本位点模型则是基于药物分子与生物靶分子之间的相互作用信息,进行基于分子力学理论及量子化学计算的药物分子设计。
三、配体基本位点亲和力模型配体基本位点亲和力模型分为静态法和动态法,静态法是通过理论计算、实验分子相互作用等方法,得到配体基本位点和生物靶分子基本位点之间的亲和力信息。
而动态法则是结合分子动力学模拟,以分子间的相互作用和运动过程,揭示配体基本位点和生物靶分子基本位点的亲和力情况。
药物设计学 第五讲QSAR
• 1863年法国斯特拉斯堡大学Cros博士的论文,他发现在哺乳动物体内 醇类的毒性随着其水溶性的降低而增加,最终可以达到一个最大值。 这是关于分子结构和生物活性之间关系的最早文献报道。
• 1868年,英国药理学家Fraser和化学家Crum Brown在研究一种生物碱 的碱性N原子甲基化前后的生物效应后,提出了化合物的生理活性依 赖于其组分的理论,即生物活性Φ应该是化合物组成C的函数: Φ=f(C) 这就是著名的Crum-Brown方程。
9
药物设计学
兰州大学药学院 李加忠
Hansch模型应用举例
Br X Y
m-X H F H Cl Cl Br I Me Br H Me H Cl Br Me Cl Me H H Me Br Br p-Y H H F H F H H H F Cl F Br Cl Cl Cl Br Br I Me Me Br Me π 0.00 0.13 0.15 0.76 0.91 0.94 1.15 0.51 1.09 0.70 0.66 1.02 1.46 1.64 1.21 1.78 1.53 1.26 0.52 1.03 1.96 1.46 σ 0.00 0.35 -0.07 0.40 0.33 0.41 0.36 -0.07 0.34 0.11 -0.14 0.15 0.51 0.52 0.04 0.55 0.08 0.14 -0.31 -0.38 0.56 0.10 Es(meta) 1.24 0.78 1.24 0.27 0.27 0.08 -0.16 0.00 0.08 1.24 0.00 1.24 0.27 0.08 0.00 0.27 0.00 1.24 1.24 0.00 0.08 0.08 log (1/C)obs 7.46 7.52 8.16 8.16 8.19 8.30 8.40 8.46 8.57 8.68 8.82 8.89 8.89 8.92 8.96 9.00 9.22 9.25 9.30 9.30 9.35 9.52 log (1/C)a 7.82 7.45 8.09 8.11 8.38 8.30 8.61 8.51 8.57 8.46 8.78 8.77 8.75 8.94 9.15 9.06 9.46 9.06 8.87 9.56 9.25 9.35 log (1/C)b 7.88 7.43 8.17 8.05 8.34 8.22 8.51 8.36 8.51 8.60 8.65 8.94 8.77 8.94 9.08 9.11 9.43 9.26 8.98 9.47 9.29 9.33
药物设计学 11第十一章 基于片段的药物分子设计

36
二、片段连接与融合
1.1 基本原理
连接 与受体结合的相邻的两个片断经连接基 连接成活性强的较大分子
37
二、片段连接与融合
1.1 基本原理
融合 与受体结合的相互交盖或接近的两个片 断合并成活性强的较大分子
38
三、片段自组装
1.1 基本原理
分别结合在活性位点中相邻口袋的两个活性片 段含有可相互反应的基团,这两个片段可自发 地反应连接成为高活性的化合物。靶蛋白在整
候选药物 HTS 苗头物 药物
uM
nM
13
生物活性
第二节 活性片段的检测技术
一、磁共振技术
基本原理
配体与生物大分子结合后,许多NMR参数(化 学位移δ)会发生改变,通过检测并分析这些数
据,可以来判定配体是否与受体结合、结合的
强弱以及结合模式。
分类
检测配体的筛选 LDBS 检测受体的筛选 TDBS
14
1. 检测配体的筛选 LDBS
1.1 原理
弛豫时间长短与分子大小成反比,小分子化合
物的弛豫时间长,大分子靶蛋白的弛豫时间短。
当药物与靶蛋白结合后就变成大分子,弛豫时
间变短。 只要适度延迟回复能量检测时间,就可以做到
只检测到游离小分子药物,而检测不到大分子
靶蛋白及其与小分子化合物的复合物。
3.1 原理
I. 通过二维15N-HSQC谱中15N或1H的化学位移变化
来检测是否有小分子与靶蛋白结合。
II. 配体和蛋白结合常数可通过化学位移的变化和配
体浓度关系测得。 III. 筛选得到结合于靶分子活性位点亚区域的低亲和
性配体,将这些配体连接可以得到具有较高亲和
药物设计学完整版

药物设计学完整版一、药物设计学概述药物设计学是一门集生物学、化学、计算机科学等多学科于一体的交叉学科,旨在通过科学的方法和技术,设计出高效、低毒、具有特定生物活性的药物分子。
药物设计学的发展,为我国新药研发提供了强有力的理论支持和实践指导。
1. 药物设计学的起源与发展药物设计学起源于20世纪50年代,随着分子生物学、计算机科学等相关学科的发展,药物设计学逐渐形成了自己的理论体系和技术方法。
经过几十年的发展,药物设计学在新药研发领域取得了举世瞩目的成果。
2. 药物设计学的主要任务药物设计学的主要任务包括:发现和验证药物作用靶点、设计具有生物活性的药物分子、优化药物分子的药效学和药代动力学性质、评估药物分子的安全性和有效性等。
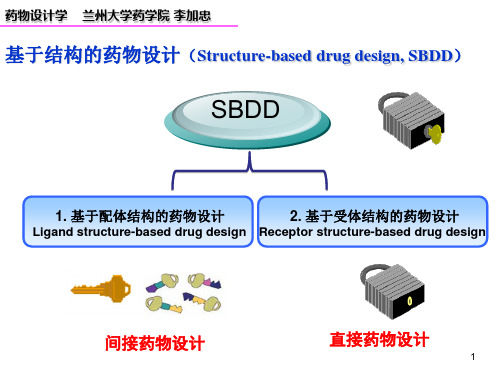
3. 药物设计学的方法与技术药物设计学的方法与技术主要包括:基于结构的药物设计、基于配体的药物设计、计算机辅助药物设计、高通量筛选等。
这些方法与技术相互补充,共同推动药物设计学的发展。
二、药物设计学的核心要素1. 靶点识别与验证药物设计的起点在于找到合适的药物作用靶点。
靶点可以是蛋白质、核酸、酶或其他生物大分子。
靶点的识别与验证是药物设计的关键步骤,它直接关系到药物设计的成功与否。
研究人员需通过生物信息学、基因敲除、基因编辑等技术手段,确保靶点与疾病的相关性。
2. 药物作用机制研究了解药物的作用机制对于药物设计至关重要。
研究人员需要探究药物分子如何与靶点相互作用,如何调控信号通路,以及如何影响疾病进程。
这有助于优化药物结构,提高药物的治疗效果。
3. 药物分子的优化三、药物设计学的应用实例1. 小分子药物设计小分子药物因其易于合成、口服给药等优点,在药物设计中占据重要地位。
例如,针对某些癌症的酪氨酸激酶抑制剂(如伊马替尼)的设计,就是基于对激酶结构的深入理解,成功开发出的靶向治疗药物。
2. 生物大分子药物设计随着生物技术的进步,生物大分子药物(如抗体、蛋白质类药物)的设计也取得了显著成果。
药物设计学(第一章先导化合物)PPT课件

先导化合物优化中的药效团模型
构效关系是研究化合物结构与活性之间关系的科学,对于先导化合物优化具有重要意义。
通过构效关系的研究,可以了解化合物中哪些结构因素对活性有影响,从而指导化合物的结构优化。
常见的构效关系研究方法包括定量构效关系(QSAR)和活性基团分析等。
先导化合物优化中的构效关系
04
先导化合物的合成与制备
基于已知的化学反应和合成路径,逐步构建目标分子的结构。
经典合成法
利用固相或液相的组合化学技术,快速生成大量结构类似的小分子库。
组合合成法
利用生物酶作为催化剂,在温和的条件下进行高效、专一的合成。
酶促合成法
先导化合物的合成方法
先导化合物的制备工艺
优化反应条件
通过调整温度、压力、溶剂等反应条件,提高目标产物的收率和纯度。
化学结构修饰、生物电子等排、拼接、类药性预测等。
03
02
01
先导化合物优化的目标与方法
药效团模型是药物设计中的一种重要工具,用于描述和预测药物与靶点之间的相互作用。
通过药效团模型,可以预测新化合物的活性,指导化合物的设计与合成,提高成功率。
药效团模型的建立需要基于已知活性化合物的数据,通过统计分析、三维构象分析等方法确定药效团特征。
特点
药物设计学的定义与特点
药物设计学是疾病治疗的重要基础,通过设计具有特定作用机制的药物,实现对疾病的预防和治疗。
疾病治疗
药物设计学是药物创新的核心环节,通过发现和设计具有新作用机制的药物,推动医药行业的创新发展。
药物创新
药物设计学的进步直接关系到人类健康水平的提高,对于保障人民生命安全具有重要意义。
人类健康
药物设计学的重要性
药物设计学(酶抑制剂含实例)

活性筛选
通过高通量筛选等方法, 找出能够与靶点结合并发 挥预期功能的候选药物。
药物设计学的研究方法
1 2
计算机辅助药物设计
利用计算机模拟技术,预测候选药物与靶点的相 互作用,从而优化药物的结构和性质。
高通量筛选
通过大规模实验筛选出具有潜在活性的候选药物, 再通过进一步验证和优化确定其药效和安全性。
物在临床上广泛应用于治疗高血压和冠心病等疾病。
05
酶抑制剂的未来发展与挑战
新药研发的挑战与机遇
挑战
新药研发过程中面临着诸多挑战,如药物靶点的确定、药物的细胞膜通透性、药 物的代谢稳定性等。同时,新药研发还需要面对临床试验的高风险和长周期。
机遇
随着科技的不断进步,新药研发的机遇也在不断增加。例如,基因组学、蛋白质 组学和结构生物学等领域的发展为药物设计提供了更多的靶点和思路。此外,计 算机辅助药物设计和高通量筛选等技术也为新药研发提供了有力支持。
酶抑制剂的作用机制
竞争性抑制剂
与酶的底物在活性位点上竞争,通过占据底物的位置来抑制酶 的活性。抑制剂与底物结构相似,抑制作用大小取决于抑制剂
与底物的亲和力。
非竞争性抑制剂
与酶的底物结合,但并不占据底物的位置,而是通过改变酶的 构象来抑制酶的活性。抑制剂与底物无竞争关系,抑制作用大
小取决于抑制剂与酶的亲和力。
实例
在抗艾滋病药物设计中,虚拟筛选技术被用于从大量小分子中筛选出针对逆转录酶的抑 制剂。
计算机辅助药物设计
总结词
计算机辅助药物设计是一种综合运用计算机技术和药学知识来设计新药物的方法。
详细描述
计算机辅助药物设计通过计算机模拟和预测药物的性质和行为,为药物设计和优化提供指 导。这种方法可以大大加速药物设计和开发的进程。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药物设计学完整版张大永骨架跃迁分子设计:从已知的活性分子结构出发,通过传统的类似物设计方法或计算化学方法,对先导化合物进行骨架设计,以发现全新的拓扑结构骨架和活性分子。
多靶点药物治疗:简而言之,可以同时作用于疾病网络中的多个靶点,对各靶点的作用可以产生协同效应,使总效应大于单个效应之和。
多靶点药物治疗可以克服许多单靶点药物的局限性,同时调节疾病网络系统中的多个环节,不易产生抗药性,达到最佳的治疗效果。
核苷类逆转录酶抑制剂(NRTI):NRTIs通过阻断病毒RNA的逆转录,即阻止病毒双链DNA形成,使病毒失去复制的模板而起作用。
它们首先进入被感染细胞,然后磷酸化,形成具有活性的三磷酸化合物。
这些三磷酸化合物是HIV逆转录酶的竞争抑制剂,当插入生长的DNA链时,可导致病毒DNA合成受阻,从而抑制病毒复制。
这类抑制剂的不良反应严重,容易使病毒产生抗药性,因此与蛋白酶抑制剂联用,常会大大延长其病毒耐药性的产生,有协同效应。
基于核酸代谢机理的药物设计:在核酸的代谢合成与代谢分解过程中,有许多酶参与其中,这些酶尤其是某些特异性的酶就成为药物设计的理想靶点;同时模拟核酸代谢过程中的底物结构,也是药物设计的一条重要途径。
核苷或核苷酸是病毒复制过程中所必需摄取的物质,通过对核苷结构的改造,可以实现对病毒复制过程的干扰。
简述具有高效耐药性NNRTIs的结构特征,并举例说明。
1. 抑制剂构象的柔性和在靶点中的可适应性理论上,在抑制剂的柔性不影响抑制剂-靶点相互作用的前提下,其构象的柔性和靶点中位置的可适应性可弥补耐药突变的不良影响,避免结合的立体位阻。
柔韧性的分子能通过化学键的自由旋转及位置的灵活移动来保持与变异靶点的紧密结合。
例:二芳基嘧啶(DAPY)类NNRTIs晶体结构表明,柔性的DAPY类分子可通过柔性扭转(摆动)和复位(微动)与不同的耐药突变的NNIBPs结合。
2.抑制剂分子与氨基酸主链形成氢键作用例:卡普韦林与p66亚基的101、103和236位残基主链间存在“网状”氢键作用。
P236主链相互作用是目前为止NNRTls的特异性作用位点。
这种多重氢键作用不受氨基酸侧链突变的影响,有助于抑制剂的自由结合,从而有效地对抗RT耐药性的产生。
因此,靶向于氨基酸残基主链的设计思想可能为对抗耐药性提供一个可靠的策略。
3.特异性靶向HIV –1 RT的高度保守性区域为了提高NNRTI的抗耐药性,应设计能与NNIBP中保守氨基酸产生特异性相互作用的抑制剂,从而降低耐药性的产生。
在NNIBP中存在一个保守性区域,主要由p225、F227、W229、L234和Y318等组成,这些氨基酸在其他慢病毒属RT中也是高度保守的。
与易突变氨基酸Y181和Y188的疏水性相互作用是第一代NNRTI的主导作用力,此外,在其它位点的突变也能通过减弱抑制剂与上述两种氨基酸相互作用而间接产生耐药性。
因此,适当降低抑制剂与Y181和Y188的相互作用有望提高化合物的抗耐药性。
例:UC-781对其它NNRTIs产生耐药性的突变株具有良好的抑制活性。
戊烯基特异性地靶向W229,由5个碳原子组成的戊烯基醚能增大UC-781与W229间的亲和力。
4.具有全新作用机制或独特结合模式的NNRTI例:苯并噻唑类衍生物CP-94707能对多个临床上常见的变异株均保持敏感性,晶体复合物研究发现,它与RT结合时呈现非常独特的结合模式。
列举三种整合酶抑制剂,并简述构效关系特点。
1. 二酮酸类抑制剂:具有有效抗病毒活性抑制剂。
作用机制:该类化合物的主要特点是相对于3′端加工,它们更偏向于抑制整合酶的链转移反应。
在链转移反应中,二酮酸类化合物与PIC中整合酶的核心结构域的两个二价金属离子活性位点相互作用,从而竞争性的抑制宿主DNA与该位点的结合。
2. 肽类抑制剂:抑制机制主要与整合酶的二聚体作用,主要代表有INH1、INH5,他们对HIV整合酶的3′端加工和链转移过程都具有明显的抑制作用。
3. 核苷类抑制剂:包括:单核苷、双核苷、三核苷及四核苷、单链、双链、三螺旋、鸟苷四聚体,它们都含有一个带负电荷的磷酸酯骨架。
主要代表AR177是一个鸟苷酸四聚体,作用机制是干扰整合酶和DNA的结合而抑制整合过程。
4. 天然来源的HIV-1整合酶抑制剂:该类抑制剂主要有黄酮和多酚,生物碱,萜类化合物,植物蛋白等。
它们来源广泛,费用低廉,副作用小。
尤其是一些来源植物蛋白已经显示出具有很好的抗病毒活性,能够协助改善AIDS的治疗。
5. 多羟基化的芳香族化合物:PHAs是整合酶抑制剂中最大的一类,大部分源于天然产物,包括:香豆素、黄酮及黄烷酮类化合物等。
它们经过化学修饰得到的衍生物具有良好的抑制整合酶的活性,大多数对整合酶有抑制作用的PHAs具有的一个共同的结构特征是分子中含有一个或多个邻苯二酚(儿茶酚)结构。
张惠斌化学库:是由诸多具有不同属性的有机化合物组成的,组合化学就是一种合成化学库的技术。
运用这项技术,不同系列的合成砌块--反应成分有序地排列起来以组成大系列的多样化分子实体群。
组合化学:是将一些基本小分子构建模块(如氨基酸、核苷酸以及各种各样的化学小分子)通过化学或者生物合成的手段,将他们系统地装配成不同的组合,由此得到大量具有结构多样性特征的分子,从而建立化学分子库的方法。
1. 固相有机合成是先把反应物接到一个固相载体(通常是官能团化的高分子材料)上,然后在非均相的条件下进行有机反应,所得最终产品用化学或者光化学法使其与作为支点的聚合物载体脱离。
反应过程简单,聚合物载体可再生,操作易实现自动化。
优点:①固相合成本身就是一种产物分离方法;②固相合成允许反应中的液相反应物大过量,因而促使固相反应物反应完全;③固相反应操作简单,因此,一系列的混合物合成方法在其基础上发展起来;④固相混合物合成可以用于超大数目的化合物库的合成。
局限性:至少一类反应物需要合适的官能团与固相载体相连接;单产物的产量有限;反应的溶剂受限制;反应过程的跟踪很复杂或根本不可能;固相反应条件的优化需要时间长。
2. 液相有机合成多组分液相反应的优化过程简单、反应速度快、操作容易、可合成的化学库体积大、产物的结构多样化程度高。
这类反应与自动化操作的结合为新药和新材料的筛选提供了及时合成化合物的可能。
但是,多组分液相反应仅局限于系列单分子合成,难于用来合成大数目的混合物,而且产物的分离往往很复杂。
创新药物:通常是指新研制的临床医疗中尚没有的药物品种,其中包括新剂型、新用途、新作用机制和新化合物,可以为临床医疗提供新的具有治疗作用的药物。
1. 改变药物应用形式的创新药物改变剂型、改变适应证、多种已知药物复方制剂等。
2. 部分创新药物指已知一定的信息资料,如作用机制、化合物的化学结构、药理作用以及临床疗效等,根据已有的药物信息,研制出的具有显著特点的新型药物。
特点:①是在药理作用方面具有突出的优势,或药效优于已有药物,或不良反应小于已有药物,或代谢特点更利于临床应用,否则这类药物则没有开发价值。
②是药物本身具有一定新颖性,所谓新颖是指有别于已知药物,或不在已知药物专利保护范围内,能够获得知识产权保护。
分类:①作用机制相同而化学结构或特点不同的物质,如钙通道阻滞剂。
②作用机制相同而且化学结构与已知药物类似新药。
如对青霉素的基本结构进行改造,发现了大批作用更强、抗菌谱更广、耐药性更小的广谱抗菌药。
③相似结构、不同作用的药物。
如阿司匹林和西地那非。
3. 完全创新药物是指在临床上至今尚没有应用的新药。
具有独特的适应证,独特的作用机制,新的作用靶点,新化学结构等。
药物筛选:就是对可能作为药用的物质进行初步药理活性的检测和试验,以求发现其药用价值和临床用途,为发展新药提供最初始的依据和资料。
常用药物筛选模型:1. 整体动物水平模型包括正常动物和病理动物模型。
优点:可以从整体水平直观地反应出药物的治疗作用,不良反应以及毒性作用。
理想的整体动物模型应具备的基本条件是病理机制与人类疾病的相似性、病理表现的稳定性和药物作用的可观察性。
缺点:主要依赖于手工操作,而且只能对有限的样品进行筛选,特别是人类目前在实验动物身上复制出的病理模型还十分有限,使用整体动物模型筛选新药具有显著的局限性、低效率和高成本等不足之处。
2. 组织器官水平模型优点:通过观察药物对特定组织或器官的作用,可以分析药物作用原理和可能具有的药理作用。
降低了筛选样品的用量;降低劳动强度,扩大筛选规模,减少动物用量;减少了影响药物作用的因素,易于评价药物作用。
缺点:主要是规模小、效率低、反应药物作用有限、对样品的需求量仍然较大,不易实现一药多筛,以及对人工操作技术要求高等。
3. 细胞分子水平模型材料用量少、药物作用机制比较明确、可实现大规模的筛选。
实现了大样本量的筛选和一药多筛。
简述药物筛选技术的发展过程,筛选的一般方法,分析现代药物筛选的优点。
发展过程:1. 原始的经验积累:无意识的自然药物筛选过程2. 神农尝百草:有意识的主动药物筛选过程3. 有目的、有计划地应用动物实验进行药物筛选4. 现代药物筛选现代科技的发展为高效率地进行药物筛选提供技术条件,促使药物筛选由整体动物实验为主转变为体外实验为主,形成了高通量药物筛选的模式,使大规模高效率的药物筛选工作在世界范围内广泛开展起来。
一般方法:动物实验法,同位素标记和检测方法,用于高通量筛选的紫外、荧光和发光检测法,计算机辅助筛选。
优点:降低了筛选样品的用量;降低劳动强度,扩大筛选规模;自动化程度高;快速、高效、灵敏。
孙宏斌酶的特异性:即酶专一性,指酶对催化的反应和反应底物所具有的选择性。
竞争性抑制剂:具有与底物相似的结构,通常与正常的底物或配体竞争酶的结合部位,形成无活性的酶-抑制剂复合物,减少酶与底物作用。
非竞争性抑制剂:底物和抑制剂与酶的结合互不相关,既不排斥,也不促进。
底物可与游离酶结合,也可和酶-抑制剂复合物结合;抑制剂可与游离酶结合,也可和酶-底物复合体结合,但酶-底物-抑制剂复合体不能释放出产物。
反竞争性抑制剂:不能与游离酶结合,但可与酶-底物复合物结合并阻止产物生成,使酶的催化活性降低。
特点:抑制剂与底物可同时与酶的不同部位结合;必须有底物存在,抑制剂才能对酶产生抑制作用。
酶的过渡态:即指底物的过渡态。
酶首先与底物结合成酶-底物复合物,然后转换成不稳定的酶-过渡态中间复合物。
过渡态与酶的亲和力要远远大于底物与酶的亲和力。
过渡态类似物的酶抑制剂:是指从电性和/或立体结构方面来模拟反应中不稳定过渡态的底物部分,该类抑制剂与酶的亲和力一般要远大于(107-105倍)底物类似物抑制剂与酶的亲和力。
举例:腺苷脱氨基酶在催化腺苷脱氨基生成肌苷的反应过程中,经过一个不稳定的水合中间体,助间型霉素和喷司他丁都是水合中间体类似物,其与腺苷脱氨基酶的亲和力比底物腺苷强106倍以上,具有免疫抑制和抗肿瘤活性。