气相色谱_质谱联用法测定人体血液中的阿托品
硫酸阿托品中莨菪碱杂质的检查例题

硫酸阿托品中莨菪碱杂质的检查例题《硫酸阿托品中莨菪碱杂质的检查例题》一、引言在制药过程中,药品的纯度和质量是至关重要的。
而作为一种常用的生物碱类药物,硫酸阿托品的纯度检查显得尤为重要。
本文将围绕硫酸阿托品中莨菪碱杂质的检查例题展开深入探讨,以期能够全面了解检查方法和意义。
二、硫酸阿托品中莨菪碱杂质的检查1. 硫酸阿托品的制备过程要了解硫酸阿托品中莨菪碱杂质的检查,首先需要了解硫酸阿托品的制备过程。
硫酸阿托品是一种高效的抗胆碱药物,其制备过程较为复杂,包括原料草药的采集、提取和精制等多个步骤。
其中,莨菪碱作为一种常见的杂质,在制备过程中很容易受到污染,因此检查其在硫酸阿托品中的含量显得尤为重要。
2. 莨菪碱的检查方法针对硫酸阿托品中莨菪碱杂质的检查,一般采用高效液相色谱法(HPLC)进行检测。
该方法具有检测灵敏度高、分辨率好等优点,能够准确测定硫酸阿托品中莨菪碱杂质的含量,并且可以进行定量分析,为制药过程提供了重要的保障。
3. 检查结果的意义莨菪碱作为一种有毒的杂质,如果其在硫酸阿托品中的含量超出标准范围,将会对药品的质量和安全性造成严重影响。
对硫酸阿托品中莨菪碱杂质的检查显得至关重要,可以有效保障药品的质量和安全性。
三、总结与展望通过以上的内容,我们对硫酸阿托品中莨菪碱杂质的检查有了更加全面和深入的理解。
在未来的制药过程中,我们需要继续加强对药品质量的监控,提高对莨菪碱杂质的检查和控制,以确保药品的质量和安全。
四、个人观点与理解作为一名制药行业的从业者,我深知药品质量对患者健康的重要性。
在制备硫酸阿托品过程中,对莨菪碱杂质的检查必须严格执行,并且要不断优化检测方法,以确保药品的纯度和质量。
只有在制药过程中严把质量关,才能为患者提供更加安全和有效的药品。
以上就是关于硫酸阿托品中莨菪碱杂质的检查例题的全面讨论,希望能够对您有所帮助。
文章总结:本文围绕硫酸阿托品中莨菪碱杂质的检查例题展开深入讨论,对药品的质量和安全性进行了全面评估。
超高效液相色谱-串联质谱法检测全血中的东莨菪碱和阿托品

血 中东莨菪碱 与阿托 品的定性定量检 测。 关键词 :东莨菪碱 ;阿托 品 ;液相 色谱 一 质谱联 用 ;快速检验
超高效液相色谱一 串联质谱法检测全血中的东莨菪碱和阿托品
佘彩 蒙 ,杜 鸿雁 ’ ,王 芳琳 2 ,何 洪源 ,王小 宝 。
( 1 . 中国人 民公 安大学 ,北京 1 0 0 0 3 8; 2 . 公安部物证鉴 定 中心 ,北京 1 0 0 0 3 8; 3 . 广 州市公安局越 秀区分局刑警 大队,广 州 5 1 0 0 8 0 )
b y u l t r a — p e r f o r ma n c e l i q u i d c h r o ma t o g r a p h y — ma s s s p e c t r o me t r y( UP L C— MS / MS ) . Hu ma n b l o o d s a mp l e s we r e p r e c i p i t a t e d
Bu r e a u , Gu a n g z h o u 5 1 0 0 8 0 ,Chi n
ABS TRACT:A n o v e l a n d r a p i d me t h o d wa s e s t a b l i s h e d f o r t h e d e t e r mi n a t i o n o f s c o p o l a mi n e a n d a  ̄o p i n e i n h u ma n b l o o d
Pe r f o r ma n c e Li q u i d Ch r o ma t o g r a p h y — Ma s s S p e c t r o me t r y
气相色谱-质谱选择离子法测定人血浆中阿拉伯糖醇DL比率

醇峰面积与"#阿拉伯糖醇峰面积之比(%)对已知的标准 !# 阿拉伯糖醇血浆浓度(&,/7/B)作直线回归,其回归方程为 %H,D"!-I8D"$$&(’H"D%%,+,(H’)。
!"",年%月 A)UT)/M)C!"",
色
谱
#$%&’(’)*+,&-.*/#$,*0-1*2,-3$4
V5*:!$G5:8 8!%
气相色谱!质谱选择离子法测定人血浆中阿拉伯糖醇 !/" 比率
张选红$, 林广云$, 蔡葵花$, 席丽艳!, 郑文晖$,
鲁长明!, 陈红英$, 汤丽芬$
($:中山大学中山医学院分析测试中心,广东 广州 8$""+";!:中山大学附属第二医院,广东 广州 8$"$!")
关键词:气相色谱#质谱;选择离子法;阿拉伯糖醇;!/" 比率;血浆
中图分类号:<’8+
文献标识码:=
文章编号:$"""#+&$,(!"",)"8#"8!%#"$
系统性念珠菌的主要代谢产物是 !#阿拉伯糖醇,因此 感染系统性念珠菌的病人血浆中 !#阿拉伯糖醇含量增高, 而其 "#阿拉伯糖醇含量几乎不变,导致阿拉伯糖醇 !/" 比 率显著增大。运用色谱技术来诊断其疾病的工作国外!"世 纪&"年代末已有报道[$],但国内仅在$%%&年有一篇有关阿 拉伯糖醇检测的报道,且其检测方法未能将 !#阿拉伯糖醇 与 "#阿拉伯糖醇分开[!]。目前国 内 尚 未 有 人 应 用 测 定 阿 拉 伯糖醇 !/" 比率的方法来诊断早期感染系统性念珠菌病。 我们利用气相色谱#质谱(>?/@A)联用技术,结合衍生化试 剂将阿拉伯糖醇的羟基卤代酰化,使极性大的阿拉伯糖醇衍
中国药品检验标准操作规范2010年版40气相色谱-质谱联用法
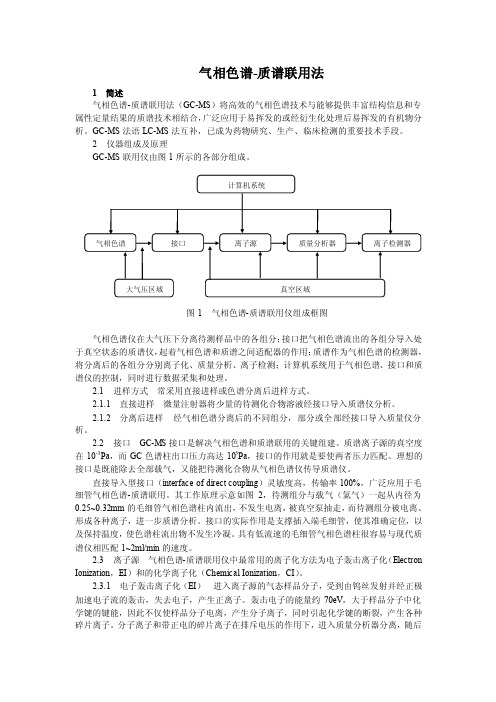
气相色谱-质谱联用法1 简述气相色谱-质谱联用法(GC-MS)将高效的气相色谱技术与能够提供丰富结构信息和专属性定量结果的质谱技术相结合,广泛应用于易挥发的或经衍生化处理后易挥发的有机物分析。
GC-MS法语LC-MS法互补,已成为药物研究、生产、临床检测的重要技术手段。
2 仪器组成及原理GC-MS联用仪由图1所示的各部分组成。
图1 气相色谱-质谱联用仪组成框图气相色谱仪在大气压下分离待测样品中的各组分;接口把气相色谱流出的各组分导入处于真空状态的质谱仪,起着气相色谱和质谱之间适配器的作用;质谱作为气相色谱的检测器,将分离后的各组分分别离子化、质量分析、离子检测;计算机系统用于气相色谱、接口和质谱仪的控制,同时进行数据采集和处理。
2.1 进样方式常采用直接进样或色谱分离后进样方式。
2.1.1 直接进样微量注射器将少量的待测化合物溶液经接口导入质谱仪分析。
2.1.2 分离后进样经气相色谱分离后的不同组分,部分或全部经接口导入质量仪分析。
2.2 接口GC-MS接口是解决气相色谱和质谱联用的关键组建。
质谱离子源的真空度在10-3Pa,而GC色谱柱出口压力高达105Pa,接口的作用就是要使两者压力匹配。
理想的接口是既能除去全部载气,又能把待测化合物从气相色谱仪传导质谱仪。
直接导入型接口(interface of direct coupling)灵敏度高,传输率100%,广泛应用于毛细管气相色谱-质谱联用。
其工作原理示意如图2,待测组分与载气(氦气)一起从内径为0.25~0.32mm的毛细管气相色谱柱内流出,不发生电离,被真空泵抽走,而待测组分被电离、形成各种离子,进一步质谱分析。
接口的实际作用是支撑插入端毛细管,使其准确定位,以及保持温度,使色谱柱流出物不发生冷凝。
具有低流速的毛细管气相色谱柱很容易与现代质谱仪相匹配1~2ml/min的速度。
2.3 离子源气相色谱-质谱联用仪中最常用的离子化方法为电子轰击离子化(Electron Ionization,EI)和的化学离子化(Chemical Ionization,CI)。
体内药物分析方法(精选)

体内药物分析方法(精选)体内药物分析方法(精选)随着现代医学的发展,药物在疾病治疗中起到了至关重要的作用。
对于新药物的研发、药物代谢的了解以及用药的个体化,需要使用合适的体内药物分析方法。
本文将介绍几种常用的体内药物分析方法。
一、液相色谱-质谱联用法(LC-MS)液相色谱-质谱联用法(LC-MS)是一种将液相色谱(LC)和质谱技术(MS)结合起来的分析方法。
它通过将待测样品进行分离,利用质谱技术对分离后的成分进行快速、准确的鉴定和定量。
LC-MS在药物代谢动力学研究、药物相互作用分析、药物残留检测、药物中间体的筛选等方面具有广泛的应用。
二、气相色谱-质谱联用法(GC-MS)气相色谱-质谱联用法(GC-MS)是一种将气相色谱(GC)和质谱技术(MS)结合起来的分析方法。
它通过将待测样品在高温条件下蒸发,然后在气相色谱柱上进行分离,最终通过质谱技术对分离后的物质进行鉴定和定量。
GC-MS在药物代谢研究、毒物学研究、药物滥用检测以及环境污染物分析等方面具有重要的应用价值。
三、原子吸收光谱法(AAS)原子吸收光谱法(AAS)是一种通过测量原子在特定波长的光束中吸收光的强度来定量分析样品中金属元素的方法。
AAS广泛用于测定药物中的微量金属元素。
例如,铁、锰、铜、锌等微量金属元素在生物体内被广泛应用。
AAS具有灵敏度高、准确性好等优点,成为体内药物分析中的重要技术手段。
四、高效液相色谱法(HPLC)高效液相色谱法(HPLC)是一种将液相色谱技术与高压技术结合起来的分析方法。
它通过将待测样品在高压下通过色谱柱进行分离,然后通过检测器对分离后的组分进行定性和定量。
HPLC广泛应用于药物代谢、药物溶出度的测定、药物杂质的分析等方面。
五、电感耦合等离子体质谱法(ICP-MS)电感耦合等离子体质谱法(ICP-MS)是一种将电感耦合等离子体技术与质谱技术结合起来的分析方法。
它利用高温等离子体对待测样品中的元素进行电离和激发,然后通过质谱技术进行分析。
气相色谱_高分辨质谱联用法检测人尿中21种兴奋剂

・
1669 ・
Target mass (m/z) 335.069 0 337.066 0 336.058 2 315.213 9 405.264 0 420.287 4 420.287 4 405.264 0 315.213 9 343.245 2 358.268 6 448.318 7 268.218 6 343.245 2 358.268 6 354.237 3 339.213 9 324.190 4 318.237 3 345.260 8 435.310 9 318.237 3 345.260 8 435.310 9 520.345 7 545.340 9 560.364 4 560.364 4 545.340 9 470.314 3 343.245 2 433.295 3 448.318 7 343.245 2 433.295 3 448.318 7 451.225 0 466.248 5 468.245 5 374.300 0 449.326 6 332.253 0 431.279 6 446.303 1 341.229 5 430.271 8 415.248 3 325.198 2 432.287 4 417.264 0 342.237 3 538.296 1 523.272 6 433.222 4 435.310 9 345.260 8 374.300 0 512.259 3 381.170 1 422.209 2 544.321 9 529.298 4 439.248 3
药学学报 Acta Pharmaceutica Sinica 2012, 47 (12): 1667−1670
・
1667 ・
气相色谱-高分辨质谱联用法检测人尿中 21 种兴奋剂
邢延一*, 刘 欣, 张玉梅, 王小兵, 徐友宣
重症监护病房急救药物的应用

某些情况下,蛋白质结合率可以发生变化, 如肾衰病人,血清蛋白质结合药物的能力下降, 游离型药物的百分比增高。因此,常规用药也 会导致药物浓度升高。严重低蛋白血症的病人, 由于血清白蛋白的降低,游离型药物也升高。 因此,临床上不能单纯根据血清药物浓度来调 整药物剂量。 三.影响药物浓度的因素 药物进入机体的量,可以通过给药量,给药 时间,给药途径等各种方式来控制。但要控制 药物从机体的排出却比较困难,因此,必须了 解影响药物排出的因素。
⑴.α受体兴奋→周围血管收缩→动脉血压升高。 ⑵.β受体兴奋→心率↑,心肌收缩力↑→心输出量↑。 ⑶.能增加心肌的应急性和自律性。 ⑷.能使细的心室纤颤变为粗的室颤,有利于除颤。
给药途径: ⑴.都倾向于中心静脉和周围大静脉给药,最 好不用下肢静脉。 ⑵.气管导管内给药显效时间与静脉一样快。
约15秒钟后血中浓度达高峰。1975年国际复苏 会议定为第二给药途径。用蒸馏水或NS稀释成 1/10000浓度,婴幼儿<5ml,成人5~10ml。 若无效,5~10min后可重复一次。 ⑶.无静脉通道时,可心内注射,剑突下给药 为好。心内注射容易引起难治性室颤、气胸、 冠脉血管破裂。以上三种给药途径至心脏复跳 时间无明显差别(139秒、132秒和127秒)。 剂量:1/10000, 0.1~0.3ml/(kg.次) iv推, 3~ 5min 可重复一次。或新生儿1ml、婴 儿 2ml、幼儿3ml、学龄前4ml、年长儿5ml。若首 次不成功,可用5~10倍的剂量(不稀释)。 持续静滴:0.01~0.05~ 0.1ug/(kg.min)
充盈压降低并增加每搏量和心排血量,从而改 善心脏功能。在改善血流动力学的同时,不增 加心肌耗氧量,不使动脉压下降是其优越性之 一。该药与洋地黄类药物联合应用具有协同作 用。此药持续静脉滴入一般可用24~48小时, 不宜长时间静脉滴入。药物半衰期一般为1小 时,但心力衰竭着半衰期可延长至2小时。 副作用可有头痛、胸痛、肌无力、震颤、 失眠、血小板减少、低血钾等。药物过量可导 致低血压,心动过速等。停药后症状一般自行 消退。肝、肾功能不全或严重心律失常着慎用。 剂量:负荷量 15~50ug/kg iv 缓推 维持量 0.25~0.75ug/(kg.min) iv负荷 后即使用
血浆中药物的浓度检测方法

血浆中药物的浓度检测方法
血浆中药物的浓度检测方法
血浆中药物的浓度检测是临床药学中的重要环节,它可以帮助医生确
定药物的剂量和疗效,从而更好地治疗患者。
目前,常用的血浆中药
物浓度检测方法主要包括高效液相色谱法、气相色谱法、质谱法等。
高效液相色谱法是一种常用的血浆中药物浓度检测方法。
它通过将血
浆样品与内标物混合后,采用高效液相色谱仪进行分离和检测。
该方
法具有分离效果好、灵敏度高、准确性高等优点,可以检测多种药物
的浓度。
气相色谱法也是一种常用的血浆中药物浓度检测方法。
它通过将血浆
样品与内标物混合后,采用气相色谱仪进行分离和检测。
该方法具有
分离效果好、灵敏度高、准确性高等优点,可以检测多种药物的浓度。
质谱法是一种高灵敏度的血浆中药物浓度检测方法。
它通过将血浆样
品与内标物混合后,采用质谱仪进行分离和检测。
该方法具有灵敏度高、准确性高等优点,可以检测多种药物的浓度。
除了以上几种方法外,还有一些新兴的血浆中药物浓度检测方法,如
电化学法、荧光法、免疫学法等。
这些方法具有灵敏度高、准确性高
等优点,但目前还处于研究阶段,需要进一步的实验验证。
总的来说,血浆中药物浓度检测是临床药学中的重要环节,不同的检
测方法各有优缺点,医生可以根据具体情况选择合适的方法进行检测。
未来,随着科技的不断发展,血浆中药物浓度检测方法也将不断更新
和完善,为临床药学的发展提供更好的支持。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
表 2 不同水解方式对提取的影响 Table 2 The influence of diff erent hydrolyzing
methods on extraction
水解方式 检测质量浓度 /( mg /L)
直接提取 0. 046
PK酶水解 强酸水解
0. 107
-
2. 5 衍生化试剂的用量和衍生化条件的选择 利用 M STFA作衍生化试剂的目的是将阿托
54
淮海工学院学报 (自然科学版 )
2004年 12月
行衍生化并定容。 进样后所得峰面积代入回归方程 进行计算 ,所得结果如表 1所示。 由表 1可以看出 , 当 pH> 9时 ,药物以原形的形式被提取到有机溶剂 中 ,故选择 p H为 10~ 11可以保证提取完全。
表 1 pH 对提取的影响 Table 1 The inf luence of pH on extraction
刘 颖 ,孙桂进 ,张炳谦
(连云港市公安局 刑警支队 ,江 苏 连云港 222002)
摘 要: 用液 -液萃取的方法提取血液中阿托品 ,经 N-甲基 -N-(三甲基硅基 )三氟乙酰胺衍生化 ,建 立了人体血液中阿托品的气相色谱 -质谱联用测定法。 结果表明 ,阿托品质量浓度在 0. 050~ 0. 40 m g / L范围内线性关系良好 ,回归方程为 A= 4. 33× 106d+ 2. 28× 103,相关系数 V为 0. 998 3,提取 回收率达 82. 1% ~ 87. 6% ,检测限达 10μg / L。 关键词: 阿托品 ;血液 ; 气质联用法 中图分类号: O657. 63 文献标识码: A
53
氯仿、异丙醇、三乙胺、氨水、盐酸等均为分析纯 ,萃 取液由氯仿、异丙醇和三乙胺按体积比 78∶ 20∶ 2 配制而成 ;蛋白酶 K( PK,美国 )。
阿托品标准品: 取硫酸阿托品对照品适量 ,用 2 m L 去离子水溶解 , 加数滴浓 氨水调节 pH= 10~ 11,用 2 m L 氯仿提取 ,分出氯仿层 ,于 60 ℃水浴上 通氮气流吹干 ,残渣备用。
验无干扰。
A— 含药血样 ; B— 空白血样 图 1 阿托品总离子流色谱 Fig. 1 The total atropine ion chromatogram
图 2 阿托品-MSTFA衍生化产物的 质谱 Fig. 2 The mass spectrum of atropine-MSTFA
2. 2 标准曲线 准确称取阿托品标准品 ,用甲醇配制质量浓度
品分子中的羟基转化为硅烷基 ,从而降低极性 ,改善 色谱行为。 由于 M ST FA试剂遇水极易分解 ,衍生 化反应要在完全隔绝水的条件下进行。 为避免少量 水对衍生化反应的影响 ,也可以加大衍生化试剂的 用量。 经过实验 ,采用 50μL衍生化试剂在 60℃反 应 30 mi n,可以保证衍生化完全 ,衍生化产物在室 温下保存 24 h ,未发生明显变化。 通过进一步实验 , 比较了阿托品衍生化前后的色谱行为 ,在给定的实 验条件下 ,保留时间没有显著变化 ,相同浓度衍生化 后的色谱峰面积比未衍生时提高了约 3倍。 2. 6 回收率及精密度试验
pH
4. 0 8. 0 9. 0 10. 0 11. 0 12. 0
质量浓度 / ( mg / L) - 0. 008 9 0. 094 0. 092 0. 086 0. 101
2. 4 水解方式的选择 由于阿托品在体内约 50% 与血浆蛋白结合 ,要
检测其浓度 ,必须将蛋白质水解 ,释放出药物 ,再进 行提取。本文以 1例阿托品中毒患者的血浆为样品 , 试验了直接提取法、酶水解法、强酸煮沸水解法对提 取的影响 ,结果如表 2所示。强酸水解因造成阿托品 本身的分解而导致无法检出 ,采用蛋白酶 K水解能 使水解完全 ,回收率较高。
称取硫酸阿托品对照品适量 ,溶于去离子水中 , 配制成相当于阿托品质量浓度为 0. 1 mg / L的工作 溶液 ,吸取 1 m L工作溶液 ,用盐酸和氨水分别调节 p H为 4. 0, 8. 0, 9. 0, 10. 0, 11. 0, 12. 0,分别加 入萃 取液 2 m L,涡旋振荡 5 mi n后离心 ,弃去上层水溶 液 ,有机层在 60℃下用氮气吹干。按“ 1. 2. 3”方法进
为 0. 05, 0. 1, 0. 2, 0. 3, 0. 4 mg / L的工作液 ,分别量 取 1 m L 工作液 ,挥干溶剂后按“ 1. 2. 3”方法进行衍 生化并定容 ,然后进样分析。 以阿托品质量浓度 (d) 为纵坐标 ,选择 m /z 为 124, 361离子作为积分离子 计算峰面积 ( A )作为横坐标 ,计算标准曲线。结果表 明 ,阿托品质量浓度在 0. 05~ 0. 4 mg / L 范围内线 性关系良好 ,回归方程为 A= 4. 33× 106d+ 2. 28× 103 ,V= 0. 998 3。 2. 3 pH对提取的影响
0. 164
82. 1
4. 6
3. 5
Hale Waihona Puke 0. 40. 350
87. 6
10. 1
8. 6
2. 7 最低检测限 在选定的试验条件下 ,当信噪比为 3时对最低
检测限进行测定 ,结果表明 ,本方法对血液中阿托品 的最低检测限为 10 μg / L。
3 结论
用拟定方法进行人体血液中阿托品的检验 ,所 得线性关系、提取回收率、精密度及最低检测限均符 合实际检测要求 ,方法灵敏、可靠。
第 13卷 第 4期 2004年 12月
淮海工学院学报 (自然科学版 ) Jo urnal o f Huaihai Institute of T echnolog y
V
o l. 13 N o. Dec. 2004
4
文章编号: 1672-6685( 2004) 04-0052-03
气相色谱 -质谱联用法测定人体血液中的阿托品
Abstract: The liquid-liquid ex t ractio n met ho d i s used t o ex tract at ropi ne f ro m hum an blood a nd
the at ropi ne i s derived by N-m ethyl-N-( t rim ethyl-sil yl) trif luo roacet ami de. In thi s case, a m ethod f o r the a nalysi s o f at ropine in hum an blo od by g as chroma tog raphy-m ass spect rom etry is produced. The cali bra tio n curv e is li ner i n t he rang e o f 0. 050~ 0. 40 m g / L of at ropine w it h cor relati on co ef fici ent Vbeing 0. 998 3 a nd the reg ression equatio n bei ng A= 4. 33× 106d+ 2. 28× 103. T he ex t ractio n recov ery rat e o f at ropi ne at di ff erent co ncent ratio ns is 82. 1% ~ 87. 6% a nd the det ecti on limit is 10μg / L. Key words: a tro pine; plasma; gas ch ro mat og raphy-mass spect romet ry
2 结果与讨论
2. 1 对比试验 同时取空白血液样品和添加阿托品后质量浓度
为 0. 1 mg / L的血液样品 ,按照“ 1. 2. 3”方法进行提 取 ,在给定 GC-M S条件下进行分析 ,所得色谱如图 1所示。 对相应的峰作扫描 ,所得质谱如图 2所示。 由图可知 ,在给定的条件下 ,空白血样对阿托品的检
Determination of Atropine in Human Plasma by Gas Chromatography-mass Spectrometry
LIU Ying , SUN Gui -ji n, Z HAN G Bi ng-qia n
( Crimina l Brigade, Lia ny ung ang Public Security Bureau, Lia ny ung ang 222002, China)
气相色谱法 [ 4]、高效液相色谱法 [ 5]、电极法 [ 6~ 7]和比 色法 [8 ]等 ,国内对人体血液中微量阿托品的分析方 法研究较少。本文运用气相色谱 -质谱联用法对人体 血液中的阿托品进行了测定
1 实验部分
1. 1 仪器和试剂 V ARIAN GC-3900 / SAT U RN-2100T GC /M S
空白血液: 购自连云港本地医院。 加氟化钠抗 凝 ,添加一定量的硫酸阿托品对照品制成含药血样。 1. 2 实验方法 1. 2. 1 色谱分析条件 V F-5M S毛细管气相色谱 柱 ,柱长 30 m ,内径 0. 25 m m,膜厚 0. 25 μm; 进样 口温度 290℃ ,柱温 100℃ ( 1 min) 10 ℃ /min 280℃ ( 10 mi n) ; 载气: He( 99. 999% ) ,恒流方 式 ,流量为 0. 8 m L /min,分 流比 100∶ 1; 瞬 间不分 流进 样方 式 ,瞬间不分流时间 0. 75 mi n。 1. 2. 2 GC-M S条件 EI离子源: 电压 70 eV ,扫描 范围 45~ 400,阱温 190℃ , 传输线温度 250℃ ,歧 管温度 40 ℃ ,灯丝电 流 10 μA, 扫描速率 0. 73 s / Scan( 3μScan) ,溶剂延迟 4 min。积分离子 124, 361。 1. 2. 3 样品提取及衍生化 取添加了阿托品标准 品的血液样品 1 m L ,加 p H为 8. 3的缓冲溶液 1. 0 m L和蛋白酶 K约 10 mg , 37℃水解过夜 ,取出放 冷 后用浓 氨水 调节 pH为 10~ 11, 加 入萃 取液 2 m L,涡旋振荡 5 mi n后离心 ,弃去上层水溶液 ,有机 溶剂用 p H为 10~ 11的氨水溶液 2 m L 洗涤两次。 分出有机层加质量分数为 10% 的盐酸溶液 2 m L, 涡旋振荡 5 mi n后离心 , 分出水层 ,用 浓氨水调节 pH为 10~ 11,加入萃取液 2 m L,涡旋振荡 5 min后 离心 ,弃去上层水溶液 ,有机层在 60 ℃下用氮气吹 干。 残渣以 50μL M ST F A分次转移至微型反应器 衬管内 ,密封后在 60 ℃下反应 30 min,取出放置 , 冷却后用氮气流在常温下吹干试剂 ,残渣用 30μL 甲醇定容后进样 ,进样量为 2μL。