有关物质方法确认方案
紫外分光光度计确认方案精选全文

可编辑修改精选全文完整版UV-1800紫外分光光度计确认方案1 概述1.1紫外-分光光度法是通过被测物质在紫外光区或可见光区的特定波长处或一定波长范围内的吸收度,对该物质进行定性和定量的方法。
本法在药品检验中主要用于药品的鉴别、检查和含量测定。
TU-1901双光束紫外可见分光光度计主要由光源、单色器、样品室、检测器等组成,波长范围为900nm-190nm,光谱带宽分6档(5,2,1,0.5,0.2,0.1),操作简便,测量快速,自动化程度高。
1.2基本情况设备名称:紫外可见分光光度计型号:生产厂家:安装日期:使用部门:质量控制部工作间:质量控制部2 确认目的确认紫外可见分光光度计测定数据准确可靠,适用于预期用途。
3 确认范围3.1 文件的适用范围本文件适用于质量控制部紫外可见分光光度计的确认。
3.2 确认的范围质量控制部紫外可见分光光度计的确认。
4 职责4.1 质量控制部职责✧负责起草确认方案、总结报告;✧负责整个确认方案的实施,并做记录、总结报告;✧负责该确认得出可靠的确认理论,适用于产品检验。
4.2 质量保证部职责✧做好过程监控,确保方案执行过程符合法规要求;4.3 动力部职责负责仪器的正常运行本仪器经实验确认,确认过程是否严格按照仪器操作规程和仪器说明书进行,试验结果稳定是否可靠,是否存在任何实验风险。
7 确认实施7.1相关文件a.仪器、仪表校验情况7.2 安装确认7.3.1 一般检查7.3.2 波长准确度以仪器中氘灯的656.1nm特征谱线检查。
在主菜单中激活光谱扫描窗口,选择【测量】菜单下的【参数设置】子菜单进行设置。
选择能量方式(Es),扫描范围(653nm-659nm),显示范围(0.0000E-50.00E)慢速扫描,采样间隔0.1nm,上述分别确认后,开始扫描。
扫描结束后按读取测得的峰值波长,其与标准波长(656.1nm)之差应不超过±7.3.3 吸光度的准确度取在120℃干燥至恒重的基准重铬酸钾约60mg ,精密称定,置1000ml 量瓶中,用0.005mol/L 硫酸溶液溶解并稀释至1000ml ,用配对的1cm 石英池,以0.005mol/L 硫酸液位空白,在235、257、313、350nm 分别测定吸光度,然后换算成%1E ,测得值应符合下表的允差范围。
方法确认与方法转移
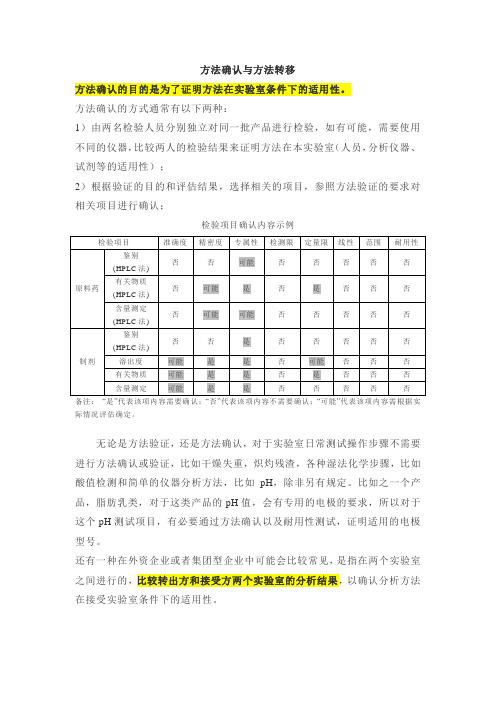
方法转移检验批次示例
类别
检验的批次数
第一类
辅料:中间产品、包装材料
原料和成品(鉴别实验或其他简单实验)
1批
第二类
原料和成品(除鉴别实验或其他简单实验外)
是
否
否
否
否
否
备注:“是”代表该项内容需要确认;“否”代表该项内容不需要确认;“可能”代表该项内容需根据实际情况评估确定。
无论是方法验证,还是方法确认,对于实验室日常测试操作步骤不需要进行方法确认或验证,比如干燥失重,炽灼残渣,各种湿法化学步骤,比如酸值检测和简单的仪器分析方法,比如pH,除非另有规定。比如之一个产品,脂肪乳类,对于这类产品的pH值,会有专用的电极的要求,所以对于这个pH测试项目,有必要通过方法确认以及耐用性测试,证明适用的电极型号。
检验项目确认内容示例
检验项目
准确度
精密度
专属性
检测限
定量限
线性
范围
耐用性
原料药
鉴别
(HPLC法)
否
否
可能
否
否
否
否
否
有关物质(HPLC法)
否
可能
是
否
是
否
否
否
含量测定(HPLC法)
否
可能
可能
否
否
否
否
否
制剂
鉴别
(HPLC法)
否
否
是
否
否
否
否
否
溶出度
可能
是
检验方法确认程序【范本模板】

一.目的:适应医学技术的发展,确保检验方法准确可靠,以满足临床需要。
二.适用范围:实验室所有开展的检验方法。
三.工作程序:1.实验室必须使用能保证准确和可靠的检验结果的检验方法、器材、仪器、试剂、质控品和校准品、供应品.2.必须选择能保证检给结果在实验室所确定的方法性能规格内的方法。
在检测标本前,实验室必须对使用方法的下述的性能规格进行确认:准确度、精密度.如有必要,可添加特异性和分析灵敏度,以及检验结果的报告范围、参考值以及其它适合的特性。
2.1.实验室引进我国注册登记的国外方法(试剂盒)或我国取得生产许可证和方法(试剂盒)时,在报告检验结果前,必须得到下列特性的性能规格:准确度、精密度、检验结果的报告范围(或者线性)、参考值。
实验室至少应检查其准确度、精密度。
必要时增加特异性和分析灵敏度,以及报告范围,参考值是否符合本实验室的服务人群等.如与厂商提供的数据进行比较,应取得相符结果。
2.2.实验室自行建立的方法,在报告结果患者前:2.2.1建立每一方法的性能规格,包括:准确度、精密度、特异性、干扰因素的影响、分析灵敏度、检验结果的报告范围(或者线性)、参考范围、其它需要检测的性能.2.2.2.建立该方法的校准程序和ICQ规则。
2.3实验室必须有上述活动的记录和文件。
并保存到停止到使用这些方法后半年。
3.实验室必须有与所做检验的专业和工作量相适应的,足够的器材、仪器、试剂、质控品和校准品、供应品。
4.实验室必须确定正确制备、储存和使用上述体外诊断用品的条件。
4.1.这些条件包括:水的质量、温度校准。
保护器材和仪器,不致由于电流的波动各中断引起对检验结果的不利影响.4.2.必须将纠正由于达不到规定所采取的改正措施文件化.5.用商品试剂盒时,对于国家规定应有生产许可证,注册登记证的品种,决不能使用没有生产许可证、注册登记证的商品试剂盒。
尚未规定者,生产厂家应提供该产品性能规格以及质量保证书。
6.试剂、溶液、培养基、校准品、质控品和其它供应品,必须加以标记,标记上应有:6.1.识别名:当有意义时应标明浓度,效价,滴度等6.2.储存要求6.3.制备日期和失效期6.4.其它与正确使用有关的信息7.应根据仪器制造商说明或依据权威机构的要求来选择和使用校准品和质控品.实验室自行选用的校准品或质控品,应有实验依据证明其不影响检验结果的准确性和可靠性。
如何进行实验室方法确认方法验证

2 方法确认的主要内容及注意事项
二、方法确认主要内容
ꢀ依据:《环境监测 分析方法标准制修订技术导则》 (HJ•168-2010)
主要内容: 检出限、精密度、准确度、 校准曲线、实际样品测试、 方法的特定要求等 ★ 可参考方法编制说明中验证方案
检出限—一般确定方法
检出限测定
要求
试验方法 含量/浓度
分析对象 测试步骤 测定方式
含有基体 样品分析的全部过程(包括前处理)
多份样品平行测定
标准溶液 直接进样 单份样品多次进样
检出限-合理性判断1(空白未检出)
判据: 要求
MDL=•t(n-1,0.99)וS, (S:标准偏差)
解决办法
单组分
比值1~10
比值>10或<1
改变样品/加标浓度
多组分
1、50%的目标物比值在3~5之间;
方法的特定要求
. 仪器性能检查-气相色谱 土壤和沉积物 有机氯农药的测定 气相色谱-质谱法土壤和沉积物 (HJ835-2017)
方法的特定要求
. 仪器性能检查-质谱 土壤和沉积物 有机氯农药的测定 气相色谱-质谱法HJ835-2017
方法的特定要求
. 仪器性能检查-质谱 土壤和沉积物 挥发性有机物的测定 吹扫捕集/气相色谱-质变样品/加标浓度;
2、至少90%的目标物比值在1~10之间;
2、相应因子差异大的
3、其余10%的目标物比值<20
组分检出限需分别测定
准确度
满足方法回收率的要求
不满足
查明原因
满足方法要求
不高于方法规定的检出限
高于方法检出限
查明原因
检出限-合理性判断2(空白有检出)
判据: 任意测定值之间允许的差异范围:
(完整版)检验方法验证和确认管理规程

页次:共11 页第1 页文件名称:检验方法验证和确认管理规程编码:03SMP01200起草审核批准颁发部门质量保证部日期日期日期实施日期分发部门及份数:质量管理部1份目的:明确检验方法的验证和确认的管理规程,确保所采用的检验方法科学、合理,符合检验要求并能有效控制药品的内在质量。
范围:仅适用于本公司对物料、产品的理化检验方法的验证和确认;清洁验证方法的验证。
职责:质量管理部QC、QA人员、质量管理部负责人对本规程的实施负责。
内容:1. 方法验证及确认工作职责分工1.1 质量控制部QC负责验证或确认方案的起草、验证或确认工作具体实施以及报告的填写。
1.2质量控制部负责人或其指定人员负责验证或确认方案、报告的审核,组织验证或确认工作的实施,对验证或确认工作中出现的问题及时纠正。
1.3 质量保证部QA负责验证或确认方案、报告的审核,监督确认工作实施,对确认工作中出现的问题提出改进意见并监督落实。
确保检验方法验证或确认程序达到符合性要求,程序被遵照执行,并且方法的预定用途被有效的且以文件记录的数据所支持。
1.4 质量管理部负责人负责验证或确认方案及报告的审核批准。
2 方法验证2.1定义:方法验证就是根据检验项目的要求,预先设置一定的验证内容和验证标准要求,并通过设计合理的实验来验证所采用的分析方法是否符合检验项目的要求。
2.2 目的:方法验证是证明采用的方法适合于相应检测要求。
2.3 适用范围:符合下列情形之一的,应当对检验方法进行验证:(1)采用新的检验方法;(2)检验方法需变更的;(3)采用《中华人民共和国药典》及其他法定标准未收载的检验方法;(4)法规规定的其他需要验证的检验方法。
文件名称:检验方法验证和确认管理规程编码:03SMP012002.3.1 在建立药品质量标准时,应对分析方法中的各检验项目进行完整的验证。
2.3.2 当药品生产工艺变更时,制剂的组分变更、原分析方法修订时,可根据变更的内容决定对分析方法进行部分验证还是完全验证。
化学检测实验室的方法验证与确认

化学检测实验室的方法验证与确认化学检验实验室主要依靠准确掌握检验方法和掌控测试结果的有效性两个方面来保障检测结果的准确性,通常对检测方法的控制要求有两个控制理念,分别为方法验证和方法确认。
一部分实验室工作人员在进行检测时,不区分标准方法和非标准方法,直接使用同一种方式和操作方法进行检测,这就会在实验室检测标准技术方面出现很大的不确定度,影响检测结果的准确性。
标签:化学检测;实验室;方法验证;确认1 方法验证和确认的定义“方法验证”对应于英文单词“Verification”,ISO对其定义为“通过检查并提供客观证据,验证规定要求得以满足”。
通过方法验证可证实方法适用于其预期目的的过程。
确认包括研究性能特征,如:准确度、特异性、检出限、定量限、線性度、范围、耐用性和稳健性。
“方法确认”对应于英文单词“Validation”,根据ISO,其定义为:“通过考察和提供客观证据,确认是否能满足某个特定预期用途的特殊要求”,即:方法确认需要“确认某个特定的分析方法能满足分析目的。
”换句话说就是“表明一个规定的方法协议,适用于某个特定类型的测试物料,以及待测物特定的浓度水平”。
通过方法确认可证实一个实验室有能力重复已确认的方法,同时方法能够达到可接受的性能水平。
从方法确认和方法验证两者的定义可知,如果一个实验室希望使用某一标准方法(已通过协作研究被全面确认),则应开展方法验证。
而某一实验室需要研发一个新方法或使用非标方法时,则应进行方法确认。
2 方法验证和确认的实施程序2.1 方法验证的实施程序一般来说,进行方法验证有以下几种情况:①采用标准方法前;②对已确认方法进行了细微改变时(例如:使用了不同厂家的相同类型的色谱柱、改变了样品稀释倍数等)。
FDA实验室手册规定,对标准方法的性能验证可分为两种情况:①批量分析情况下的性能验证;②实验室首次使用标准方法前的验证。
而前者更多意义上是属于内部质量控制的范畴。
在方法验证过程中,需要考虑哪些关键参数,取决于方法的性质和可能遇到的样品类型。
化学分析方法验证

线性
线性是指再设计范围内,测试结果与试样中被 测物浓度直接呈正比例关系的程度。 线性应在分析需要的范围内进行评价 一般以最小二乘法处理数据求的回归曲线的斜 率来表示,必要时可对响应信号进行数字转换。 数据要求:至少需要五个浓度考察线性,需提 供相关系数,Y截距,应列出回归方度,准确度和线性条件下,测 试方法是用的高,低限量浓度或量的区间。 范围应根据分析方法的具体应用和线性,准确度,精 密度结果和要求确定。 原料药和制剂含量测定范围为80%-120% 制剂含量均匀度范围为70%-130% 杂质测定应为被测杂质限度的-20%到+20% 溶出度应为测定范围的-20%到+20%,释放度因规定 了限度范围,应为下限的-20%到上线的+20%,例如 缓释片1h<20%,7h>70%,则验证范围定为0-90%
化学分析方法验证
化学分许方法验证成功的前提
能够研究建立一个有水准的分析方法的能力
有化学分析方法验证的知识和水平
化学分许方法验证成功的前提
有水平,经过培训的实施人员 仪器性能已经得到确认(校准,验证,校准在 有效期内) 可靠的对照品(法定机构提供的或有水平自己 建立的) 质量可靠的实验试剂
SD 100% x
精密度
重复性:在相同条件下,有同一个分析人员测定所得 结果的精密度
在规定的范围内,至少用9次测定结果评价,如制备 不同浓度样 品各测三次 把被测浓度当做100%,至少测6次进行评价
中间精密度:同一实验室,不同时间有不同的分析人 员用不同设备所得结果的精密度(三人三机) 重现性:不同实验室,不同分析人员测定结果的精密 度。以SD,RSD表示。
药典方法确认规程
目的建立药典方法确认规范,确保药典方法适用于本实验室的实验条件。
定义药典方法的确认:收载于CP、USP、BP、EP等药典的产品分析方法,使用前证明该验证过的产品测试方法、条件确实适用于本实验室的实际使用环境。
职责质量控制室所有人员负责按照本规程执行药典分析方法的确认。
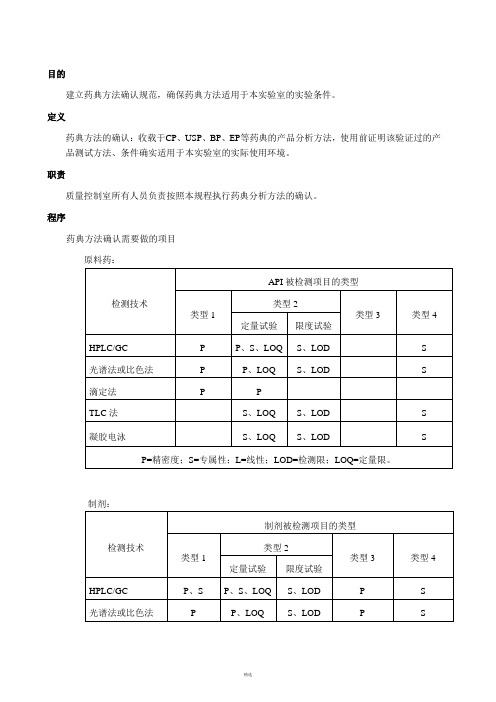
程序药典方法确认需要做的项目原料药:制剂:被检测项目类型的说明:系统适用性实验按药典正文要求的内容进行;正文没有要求的内容,如RSD%,按药典附录的要求进行。
专属性系指在其他成分(如杂质、降解物、辅料等)可能存在下,采用的分析方法能够正确鉴定、检出被分析物质的特性。
通常,鉴别、杂质检查、含量测定方法中均应考察其专属性。
如采用的方法不够专属,应采用多个方法予以补充。
鉴别反应专属性鉴别试验应确证被分析物符合其特征。
专属性试验要求证明能与可能共存的物质或结构相似化合物区分,需确证含被分析物的供试品呈正反应,而不含被测成分的阴性对照呈负反应,结构相似或组分中的有关化合物也应呈负反应。
杂质检测专属性在杂质可获得的情况下,可向供试品中加入一定量的杂质,证明杂质与共存物质能得到分离和检出,并具适当的准确度与精密度。
在杂质或降解产物不能获得的情况下,专属性可通过与另一种已证明合理但分离或检测原理不同、或具较强分辨能力的方法进行结果比较来确定。
或将供试品用强光照射,高温,高湿,酸、碱水解及氧化的方法进行破坏(制剂应考虑辅料的影响),比较破坏前后检出的杂质个数和量。
必要时可采用二极管阵列检测和质谱检测,进行色谱峰纯度检查。
含量检测专属性➢对于API主成分含量测定可在供试品中加入杂质,考察测定结果是否受干扰,并与未加杂质的供试品比较测定结果;在杂质或降解产物不能获得的情况下,可采用另一个经验证了的或药典方法进行比较,对比两种方法测定的结果。
也可采用破坏性试验(强光照射,高温,高湿,酸、碱水解及氧化),得到含有杂质或降解产物的试样,用两种方法进行含量测定,比较测定结果。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
METHOD VERIFICATION PROTOCOL FOR CHROMATOGRAPHIC PURITY OF CYTARABINE 阿糖胞苷有关物质检查方法确认方案Area to be Distributed:AD, QA分发部门:分析部,质量保证部Print Name:姓名:Title:职位:Signature:签名:Date:日期:Author:起草人:Approver:批准人:Approver:批准人:Reviewer:审核人:Approver:批准人:Contents目录一、Introduction (3)二、Scope (3)三、Responsibility (3)四、Definitions (3)五、Project (5)六、References (25)一、Introduction简介## is in the process of developing Cytarabine Injection, a liquid product for parenteraladministration, for ##. Cytarabine is a chemotherapy agent with a molecular weight of243.2. The API from Zhejiang Hisun Pharmaceutical Co. Ltd, which is approved by FDA,is used for Cytarabine Injection by Lummy.The Chromatographic purity method forCytarabine is the method in the current USP34. The HPLC method for Chromatographicpurity test of Cytarabine is a quantitative method. The method will be verified accordingto USP<1226>, including system suitability, specificity, LOD, LOQ, precision, solutionstability and mobile phase stability.阿糖胞苷注射液是一种临床用液体产品,##药业为##药业研发该品种。
阿糖胞苷是一种化学药,分子量为243.2。
莱美研发阿糖胞苷注射液所用原料药来源于浙江海正药业,是获得了FDA认证的原料药。
阿糖胞苷色谱纯度检测方法来自现行USP34,色谱纯度检查方法,是定量检测方法。
依照USP<1226>,本次确认内容包括系统适用性,专属性,检测限,定量限、精密度、溶液稳定性和流动相稳定性。
二、Scope范围This protocol applies to the verification for chromatographic purity of cytarabine.该方案适用于阿糖胞苷色谱纯度的方法确认。
三、Responsibility职责Name 姓名Department所在部门Title职称/职务Responsibility职责Group leader, responsible for approveof protocols, records and reports, andorganize the implementation of theprotocol.组长,负责方案、记录和报告的审查批准并组织方案的实施。
Initiate and implement the protocol起草并执行方案Approve protocols, records and reports方案、记录和报告的审查批准Monitor the implementation of theprotocol监督确认方案的实施四、Definitions定义1 Limit of Detection (LOD)检测限(LOD)The limit of detection of an individual analytical procedure is the lowest amount of analytein a sample which can be detected but not necessarily quantitated as an exact value.分析规程中的检测限是指样品中的被分析物在该分析方法中能被检测,但不需要准确定量的最低浓度。
The detection limit is determined by analyzing samples with known concentrations ofanalyte and by establishing the minimum level at which the analyte can be reliablydetected.检测限是通过分析样品已知浓度的被分析物,从而建立其可稳定检测的最低浓度水平。
2 Limit of Quantitation(LOQ)定量限(LOQ)The quantitation limit of an individual analytical procedure is the lowest amount ofanalyte in a sample which can be quantitatively determined with suitable precision andaccuracy.分析方法中的定量限是指样品中被分析物能够被定量检测并保证一定精密度和准确度的最低浓度。
The limit of quantitation is a parameter of quantitative assays for low levels of compoundsin sample matrices, and is used particularly for the determination of impurities and/ordegradation products.定量限是在样品中低浓度化合物被定量检测的参数,该参数经常被用于杂质和/或降解产物的测定。
3 system suitability系统适用性System suitability tests are an integral part of gas and liquid chromatographic methods.They are used to verify that resolution and reproducibility of the chromatographic systemare adequate for the analysis to be done. The tests are based on the concept that theequipment, electronics, analytical operations, and sample to be analyzed constitute anintegral system that can be evaluated as such.系统适用性试验是气相色谱和液相色谱方法的必要组成部。
用以确认该分析方法色谱系统的分离度和重现性是适用的。
该试验把分析设备、电子仪器、分析操作和分析样品作为一个整体来进行评估。
4 Specificity专属性Specificity is the ability to assess the analyte unequivocally in the presence of componentswhich may be expected to be present. Typically these might include process impurities,degradates, matrix, etc.专属性是指在一些预期组分的存在下,被分析物被明确检测评估的能力。
通常这些预期组分是指工艺杂质,降解产物,基质等。
5 Precision精密度The precision of an analytical procedure expresses the closeness of agreement (degree ofscatter) between a series of measurements obtained from multiple sampling of the samehomogeneous sample under the prescribed conditions.分析过程的精密度是指同一样品在指定的条件下所做的一系列测试值的相近程度(离散度)。
5.1 Repeatability重复性Repeatability expresses the precision under the same operating conditions over a shortinterval of time.重复性是指同一个实验室相同操作条件下短期内测定实验结果的精密度。
5.2 Intermediate precision中间精密度Intermediate precision expresses within-laboratories variations: different days, differentanalysts, different equipment, etc.中间精密度是指在同一实验室内改变其他条件,包括不同的日期,不同的人,不同的仪器等的精密度。
五、Project方案内容1 Acceptance criteria可接受标准Items 确认参数Acceptance Criteria 可接受标准System suitability 系统适用性Six replicate injections of Standard solution, RSD ≤3.0%.重复进样标准溶液6次,RSD ≤3.0%Resolution≥1.25 (between cytarabine and uridine)阿糖胞苷与尿苷分离度≥1.25The S/N ratio of the Cytarabine peak and the noise near theCytarabine in the System sensitivity solution is not less than 10.系统灵敏度溶液中阿糖胞苷峰与其附近噪音的信噪比不小于10。