新旧申报资料2.3.S 原料药汇总表对比
CTD格式申报资料撰写要求内容

国食药监注〔 2010〕 387 号附件:化学药品 CTD格式申报资料撰写要求CTD格式申报主要研究信息汇总表(原料药)2.3.S.1基本信息2.3.S.1.1药品名称原料药的中英文通用名、化学名2.3.S.1.2结构原料药的结构式、分子式、分子量2.3.S.1.3理化性质原料药的主要物理和化学性质:性状(如外观,颜色,物理状态);熔点或沸点;比旋度,溶解性,溶液pH, 分配系数,解离常数,将用于制剂生产的物理形态(如多晶型、溶剂化物、或水合物),生物学活性等。
2.3.S.2生产信息2.3.S.2.1生产商生产商的名称(一定要写全称)、地址以及生产场所的地址。
2.3.S.2.2生产工艺和过程控制(1)工艺流程图:参见申报资料3.2.S.2.2 (注明页码)。
(2)工艺描述:按反应路线简述各步反应的反应类型(氧化、还原、取代、缩合、烃化、酰化等),各步反应的原料、试剂、溶剂和产物的名称,终产物的精制方法和粒度控制等;特殊的反应条件(如高温、高压、深冷等)应说明。
详细容参见申报资料3.2.S.2.2 (注明页码)。
(3)生产设备:参见申报资料3.2.S.2.2 (注明页码)。
(4)大生产的拟定批量:kg(g)/ 批。
2.3.S.2.3物料控制生产用物料(如起始物料、反应试剂、溶剂、催化剂等)的质量控制信息(包括来源、质量标准等),参见申报资料3.2.S.2.3 (注明页码)。
2.3.S.2.4关键步骤和中间体的控制列出所有关键步骤及其工艺参数控制围。
关键步骤确定依据参见申报资料中间体的质量控制参见申报资料3.2.S.2.4 或3.2.S.2.6 (注明页码)。
3.2.S.2.4 (注明页码)。
2.3.S.2.5 工艺验证和评价无菌原料药:工艺验证方案(编号:-- ,版本号:-- )和验证报告(编-- ,版本号:--参见申报资料3.2.S.2.5 (注明页码)。
其他原料药:工艺验证方案(编号:-- ,版本号:-- )和验证报告(编-- ,版本号:--参见申报资料3.2.S.2.5 (注明页;或者,工艺验证方案(编号:-- ,版本-- )和批生产录(编号:-- ,版本号:-- )样稿参见申报资料3.2.S.2.5 (注明页码),验证承诺书参见申报资料3.2.S.2.5(注明页码)。
药品申报三类与四类药比较

新注册分类3类与4类原料药申报资料要求比较
国家局近日公布了《化学药品注册分类改革工作方案(征求意见稿)》,其中药品注册分类大家之前均已有了解,此次征求意见稿并无大的变化。
不过有两点内容应引起关注,一是监测期的变化,二是该方案中对于3类、4类申报资料的要求的区别。
下面就3类与4类原料药申报资料的区别进行汇总如下:
(1)3类申报资料要求与387号文一致,没有变化。
而4类申报资料的要求应该是新撰写修订的,其中的许多要求更加具体、明确,便于研究人员理解操作,支持!
(2)4类申报资料要求中多次提到了参考ICH相关指导原则,说明国内的研发与审评更加的与国际接轨了,研发人员更应该好好的研读ICH的相关指导原则。
(3)4类申报资料要求中多次提到了杂质研究,可见目前仿制药(特别是原料药)杂质研究仍然是高度关注的重点。
(4)4类申报资料要求中明确了一些概念,并且与目前国内的核查要求衔接更加紧密,例如关键起始物料的供应商审计,应引起大家的重视。
(5)4类中明确要求申报时需要提交12个月的长期稳定性试验数据,如此一来,国内仿制药研发的进度又会减慢了。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23。
20160504-2016年第80号通告附件-原料药CTD格式药学研究信息汇总表

附件1原料药CTD格式药学研究信息汇总表2.3.S.1基本信息2.3.S.1.1药品名称列出原料药的中英文通用名、化学名,并应与中国药典或国内已上市产品保持一致。
2.3.S.1.2结构列出原料药的结构式,并应与中国药典或国内已上市产品保持一致。
2.3.S.1.3理化性质简述本品的主要理化性质及数据来源。
2.3.S.2生产信息2.3.S.2.1生产商提供生产商的名称(全称)、地址、电话、传真以及生产线的具体地址、电话、传真等。
2.3.S.2.2生产工艺和过程控制(1)反应式:对于化学合成生产的原料药应提供反应方程式,明确所用的试剂、溶剂、催化剂与反应条件。
对于提取或发酵等工艺生产的原料药,应提供工艺流程图,明确所用的试剂、溶剂与主要工艺参数。
(2)工艺描述:按工艺路线简述各步反应的反应类型(如,氧化、还原、取代、缩合、烃化、酰化等)。
以目前生产的最大批量为例,按照各生产工序的先后简述各步反应的原料、试剂、溶剂和产物的名称、投料量(重量、摩尔比)、反应条件(温度、时间等)、各中间体的重量与收率,终产物的精制方法和粒度控制等。
(3)生产设备:列表提供本品实际生产线主要生产设备的相关信息,如型号、材质、操作原理、正常的批量范围、生产厂、用于的反应步骤等,并说明与现有最大批量的匹配性。
如不匹配,应提供充分的试验依据。
(4)大生产的拟定批量(如100kg/批)及依据:说明大生产的批量及其制定依据。
如拟定的批量超出了目前生产的最大批量所用生产设备的正常批量范围,应提供放大研究的依据。
2.3.S.2.3物料控制简述主要生产用物料(如起始物料、催化剂等)的来源、质量控制项目与限度等。
简述起始物料选择确定的合理性依据。
对于外购的起始物料,为避免对原料药的质量引入不可控因素,应提供生产商出具的制备工艺,并根据其对后续工艺的影响制订起始物料的内控标准、质检报告,说明内控标准(尤其是杂质限度与含量)的制定依据。
简述关键起始物料供应商审计计划和审计报告。
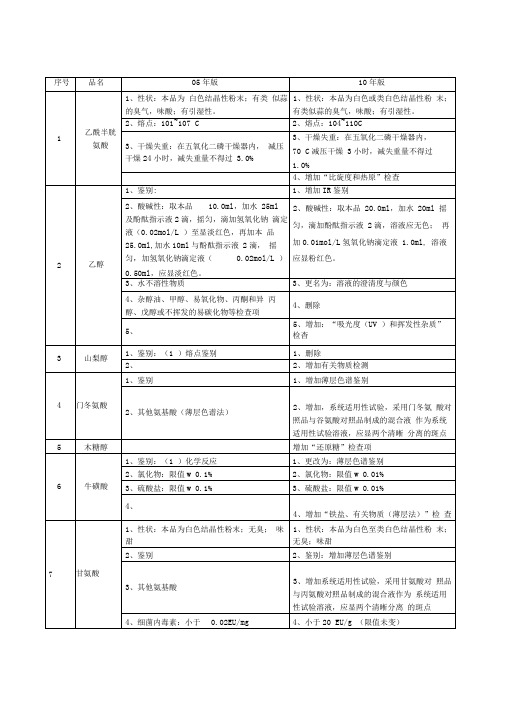
新旧药典原辅料对比

取10μl,注入液相色谱仪,盐酸丁咯地尔峰与苯甲酸峰的分离度应大于4.0。
3
山梨醇
1、鉴别:(1)熔点鉴别
1、删除
2、
2、增加有关物质检测
4
门冬氨酸
1、鉴别
1、增加薄层色谱鉴别
2、其他氨基酸(薄层色谱法)
2、增加,系统适用性试验,采用门冬氨酸对照品与谷氨酸对照品制成的混合液作为系统适用性试验溶液,应显两个清晰分离的斑点
5
木糖醇
增加“还原糖”检查项
6
牛磺酸
1、鉴别:(1)化学反应
原辅料对比
序号
品名
05年版
10年版
1
乙酰半胱氨酸
1、性状:本品为白色结晶性粉末;有类似蒜的臭气,味酸;有引湿性。
1、性状:本品为白色或类白色结晶性粉末;有类似蒜的臭气,味酸;有引湿性。
2、熔点:101~107℃
2、熔点:104~110℃
3、干燥失重:在五氧化二磷干燥器内,减压干燥24小时,减失重量不得过3.0%
对照溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分峰的峰高约为记录仪满量程的10%,再精密量取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍,供试品溶液的色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积的0.5倍(0.25%;)各杂质峰面积的和不得大于对照溶液主峰的面积(0.5%)。
1、溶解度:本品在热水中溶解,在水中微溶,在乙醇、丙酮或乙醚中不溶,在稀盐酸或1mol/L氢氧化钠溶液中易溶
药品申报三类与四类药比较
新注册分类3类与4类原料药申报资料要求比较
国家局近日公布了《化学药品注册分类改革工作方案(征求意见稿)》,其中药品注册分类大家之前均已有了解,此次征求意见稿并无大的变化。
不过有两点内容应引起关注,一是监测期的变化,二是该方案中对于3类、4类申报资料的要求的区别。
下面就3类与4类原料药申报资料的区别进行汇总如下:
(1)3类申报资料要求与387号文一致,没有变化。
而4类申报资料的要求应该是新撰写修订的,其中的许多要求更加具体、明确,便于研究人员理解操作,支持!
(2)4类申报资料要求中多次提到了参考ICH相关指导原则,说明国内的研发与审评更加的与国际接轨了,研发人员更应该好好的研读ICH的相关指导原则。
(3)4类申报资料要求中多次提到了杂质研究,可见目前仿制药(特别是原料药)杂质研究仍然是高度关注的重点。
(4)4类申报资料要求中明确了一些概念,并且与目前国内的核查要求衔接更加紧密,例如关键起始物料的供应商审计,应引起大家的重视。
(5)4类中明确要求申报时需要提交12个月的长期稳定性试验数据,如此一来,国内仿制药研发的进度又会减慢了。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23。
新注册分类3类与4类原料药申报资料要求比较

新注册分类3类与4类原料药申报资料要求比较
国家局近日公布了《化学药品注册分类改革工作方案(征求意见稿)》,其中药品注册分类大家之前均已有了解,此次征求意见稿并无大的变化。
不过有两点内容应引起关注,一是监测期的变化,二是该方案中对于3类、4类申报资料的要求的区别。
下面就3类与4类原料药申报资料的区别进行汇总如下:
(1)3类申报资料要求与387号文一致,没有变化。
而4类申报资料的要求应该是新撰写修订的,其中的许多要求更加具体、明确,便于研究人员理解操作,支持!
(2)4类申报资料要求中多次提到了参考ICH相关指导原则,说明国内的研发与审评更加的与国际接轨了,研发人员更应该好好的研读ICH的相关指导原则。
(3)4类申报资料要求中多次提到了杂质研究,可见目前仿制药(特别是原料药)杂质研究仍然是高度关注的重点。
(4)4类申报资料要求中明确了一些概念,并且与目前国内的核查要求衔接更加紧密,例如关键起始物料的供应商审计,应引起大家的重视。
(5)4类中明确要求申报时需要提交12个月的长期稳定性试验数据,如此一来,国内仿制药研发的进度又会减慢了。
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全。
原料药 80号文登记资料 附件1(1)

附件1原料药CTD格式药学研究信息汇总表2.3.S.1 基本信息2.3.S.1.1 药品名称列出原料药的中英文通用名、化学名,并应与中国药典或国内已上市产品保持一致。
2.3.S.1.2 结构列出原料药的结构式,并应与中国药典或国内已上市产品保持一致。
2.3.S.1.3 理化性质简述本品的主要理化性质及数据来源。
2.3.S.2 生产信息2.3.S.2.1 生产商提供生产商的名称(全称)、地址、电话、传真以及生产线的具体地址、电话、传真等。
2.3.S.2.2 生产工艺和过程控制(1)反应式:对于化学合成生产的原料药应提供反应方程式,明确所用的试剂、溶剂、催化剂与反应条件。
对于提取或发酵等工艺生产的原料药,应提供工艺流程图,明确所用的试剂、溶剂与主要工艺参数。
(2)工艺描述:按工艺路线简述各步反应的反应类型(如,氧化、还原、取代、缩合、烃化、酰化等)。
以目前生产的最大批量为例,按照各生产工序的先后简述各步反应的原料、试剂、溶剂和产物的名称、投料量(重量、摩尔比)、反应条件(温度、时间等)、各中间体的重量与收率,终产物的精制方法和粒度控制等。
(3)生产设备:列表提供本品实际生产线主要生产设备的相关信息,如型号、材质、操作原理、正常的批量范围、生产厂、用于的反应步骤等,并说明与现有最大批量的匹配性。
如不匹配,应提供充分的试验依据。
(4)大生产的拟定批量(如100kg/批)及依据:说明大生产的批量及其制定依据。
如拟定的批量超出了目前生产的最大批量所用生产设备的正常批量范围,应提供放大研究的依据。
2.3.S.2.3 物料控制简述主要生产用物料(如起始物料、催化剂等)的来源、质量控制项目与限度等。
简述起始物料选择确定的合理性依据。
对于外购的起始物料,为避免对原料药的质量引入不可控因素,应提供生产商出具的制备工艺,并根据其对后续工艺的影响制订起始物料的内控标准、质检报告,说明内控标准(尤其是杂质限度与含量)的制定依据。
新旧药物典原辅料对比
4、更名为“有关物质”:硝酸异山梨酯 与2—单硝酸异山梨酯按外标法计算,均
不得过0.25%;其他单个杂质峰面积不得 大于对照溶液中单硝酸异山梨酯峰面积
的0.5倍(0.25%),杂质总量不得过0.5%°
5、炽灼残渣
删除
27
注射用水
1、pH值:取本品检测
1、pH值:取本品100ml,加饱和氯化钾
2、鉴别
2、增加薄层色谱鉴别
3、其他氨基酸
3、增加系统适用性试验,采用色氨酸对 照品与酪氨酸对照品制成的混合液作为 系统适用性试验溶液,应显两个清晰分离 的斑点
15
异亮氨酸
1、鉴别
1、增加薄层色谱鉴别
2、其他氨基酸
2、增加系统适用性试验,采用异亮氨酸 对照品与缬氨酸对照品制成的混合液作 为系统适用性试验溶液,应显两个清晰分 离的斑点
与红霉素峰的分离度应不小于15.0,罗红
霉素峰与相对保留时间约为0.95处杂质
峰的分离度应不小于1.0,与相对保留时
间约为1.2处杂质峰的分离度不小于2.0°
24
乳酸
1、含量:应为85.0~90.0%(g/g)
1、含量:应为85.0~92.0%(g/g)
2、性状:本品为无色至微黄色的澄清黏 性液体;几乎无臭,味微酸;有引湿性,
5
木糖醇
增加“还原糖”检查项
6
牛磺酸
1、鉴别:(1)化学反应
1、更改为:薄层色谱鉴别
2、氯化物:限值w0.1%
2、氯化物:限值w0.01%
3、硫酸盐:限值w0.1%
3、硫酸盐:限值w0.01%
4、
4、增加“铁盐、有关物质(薄层法)”检 查
7
甘氨酸
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2.3.S.2.1 生产商
新旧版资料一致
简述主要生产用物料(如起始物料、催化剂等)的来源、质量控制项目与限度等。
对于外购的、离终产品仅三步化学反应以内的起始物料,为避免对原料药的质量引入不可控因素,应提供生产商出具的制备工艺、内控质量标准、质检报告,并根据其对后续工艺的影响制订原料药厂的内控标准,说明内控标准(尤其是杂质限度与含量)的制定依据。
简述关键的起始物料的供应商审计要求。
2.3.S.2.4 关键步骤和中间体的控制
新旧版资料大致一致。
2.3.S.2.5 工艺验证和评价
无菌原料药:要求简述工艺验证报告的主要内容:验证的时间、地点、批次、批量、验证的主要内容(关键工艺参数的验证情况),具体的生产线及主要设备,验证的结论。
其他原料药:要求简述工艺验证方案的主要内容:拟验证的时间、地点、批次、批量、拟验证的主要内容(关键工艺参数的验证情况),具体的生产线及主要设备,验证的可接受标准。
以附件形式提供验证承诺书及空白的批生产记录样稿(应与今后正常生产本品的SOP保持一致)。
2.3.S.2.6 生产工艺的开发
简述在开发阶段对哪些工艺步骤以何质量指标进行了工艺条件的优选与放大研究,并提供相关的综述资料与实验数据的汇总表。
简述工艺开发过程中生产工艺的主要变化(包括批量、设备、关键工艺参数以及工艺路线等的变化),可列表描述(示例见下表)。
并要求列表说明工艺开发
新版标题改为杂质谱分析
结合起始原料可能引入的杂质、原料药的制备工艺(中间体、副产物)、降解产物等,对可能存在的杂质进行全面的分析和研究。
质所进行的分析和研究结果,并制定合理的控制策略。
明确各杂质对照品的来源、批号、纯度等信息。
2.3.S.4 原料药的质量控制
2.3.S.4.1 质量标准
新版要求与其他药典标准进行对比,如下:
新版资料要求对其他药典收载的主要项目(如,有关物质、异构体、含量)的方法列表进行比较,示例如下:
杂质分析方法的验证应采用拟控制的杂质的对照品进行验证,以充分证明该分析方法确实能有效地检出相应的杂质。
方法学验证总结表格增加一列可接受标准。
2.3.S.4.4 批检验报告
一致
2.3.S.4.5 质量标准制定依据
简述质量标准制定依据:包括各检测项目的是否纳入质量标准的依据、限度的制定依据。
提供本品与已上市原研发厂生产的原料药(或制剂)的质量对比资料,可列表比较。
示例如下:
些杂质进行进行必要的定性研究,以证明其与原研厂产品中所含杂质的结构是一致的,且杂质含量不高于原研厂产品。
:2.3.S.5 对照品
新版要求如使用自制对照品,应提供详细的制备方法、结构研究、含量和纯度标定过程。
2.3.S.6 包装材料和容器
一致
2.3.S.7 稳定性
2.3.S.7.1 稳定性总结
一致
2.3.S.7.2 上市后稳定性承诺和稳定性方案
一致
2.3.S.7.3 稳定性数据汇总
应明确长期留样稳定性考察中是否有超过鉴定限度的杂质,并对其进行必要的定性研究,以证明其与原研厂产品中所含杂质的结构是一致的,且杂质含量不高于原研厂近效期的产品。
说明:正文标题采用宋体四号,正文为宋体小四号,1.5倍行间距,表格采用竖版,宋体五号,单倍行间距。