核磁溶剂化学位移
nmr的横坐标化学位移

核磁共振(Nuclear Magnetic Resonance,NMR)中的横坐标通常是化学位移(Chemical Shift)。
化学位移是描述核磁共振谱中峰值位置的参数,用来表示化学物质中核磁共振信号与参比物质信号之间的相对偏移程度。
化学位移的值通常以δ(delta)表示,并以部分百万(ppm)为单位。
具体说,化学位移的计算公式如下:
δ= (v - v_ref) / v_ref
其中,δ为化学位移,v为待测样品的共振频率,v_ref为参比物质的共振频率。
参比物质通常是一种具有明确化学位移的标准物质,其共振频率在不同实验条件下相对稳定。
化学位移的数值与多种因素相关,包括分子环境、化学键类型、电子密度等。
不同的化学官能团和原子类型通常具有特定范围的化学位移值,这使得化学位移成为NMR谱图解析中的重要信息。
需要注意的是,化学位移的数值对比仅在相同实验条件下具有意义,因此在NMR实验和数据解读中,通常需要参照相同仪器、溶剂和实验条件下的谱图或文献数据进行分析和比
对。
溶剂峰化学位移表__解释说明以及概述

溶剂峰化学位移表解释说明以及概述1. 引言1.1 概述溶剂峰化学位移表是有机化学中一个重要的参考工具,用于研究和识别在核磁共振(NMR)光谱中出现的溶剂峰。
在有机合成和结构鉴定等领域,正确解读溶剂峰化学位移对准确确定分子结构起着至关重要的作用。
因此,我们需要深入了解并掌握溶剂峰化学位移表的使用方法。
1.2 文章结构本文将从以下几个方面对溶剂峰化学位移表进行解释说明和概述:- 第2部分将解释定义溶剂峰化学位移,以及影响其数值的因素,并介绍计算这一参数的方法。
- 第3部分将详细阐述如何使用溶剂峰化学位移表,并提供一些范例解析。
同时,还将介绍在有机合成中应用该数据的实际案例,以及常见溶剂的峰化学位移数据。
- 第4部分将讨论溶剂峰化学位移表存在的局限性和问题,并提出改进方法来提高其可靠性和应用价值。
- 最后,在第5部分中我们将对全文进行总结,并展望溶剂峰化学位移表的发展和应用前景。
1.3 目的本文的目的是帮助读者深入理解溶剂峰化学位移表,并掌握准确使用该表以解读NMR光谱中的溶剂峰。
同时,我们还将讨论溶剂峰化学位移表存在的局限性,并提出改进方法来提高其可靠性与应用价值。
期望本文能对有机化学研究者、有机合成领域从业人员以及相关科研工作者提供有益的参考信息。
2. 溶剂峰化学位移表的解释说明2.1 溶剂峰化学位移的定义溶剂峰化学位移是指在核磁共振(NMR)光谱中,由于不同溶剂对样品的影响而导致的化学位移变化。
每种溶剂都会产生一个特定的峰,其化学位移值可以作为溶剂信号进行测量和分析。
2.2 影响溶剂峰化学位移的因素溶剂峰化学位移受多种因素影响,包括物理性质、磁性等。
其中一些可能影响峰位置和强度以及NMR光谱形状的因素包括普透明度、离子强度、极性、粘度、重金属离子污染等。
2.3 计算溶剂峰化学位移的方法计算溶剂峰化学位移常使用参考标准物质与待测样品进行比较。
通过测量已知标准物质并确定其相对于参考峰位置的差距,可以计算待测样品中各个组分的相对位置。
溶剂峰化学位移

溶剂峰化学位移
溶剂峰化学位移(Solvent Peak Chemical Shift,简称SPCS)是指在核磁共振实验中发生的现象,它表明溶剂对核磁共振信号的影响可以产生有意义的化学位移。
由于溶剂对基团的极化作用的存在,基团原子的化学位置会发生变化,导致核磁共振信号随着溶剂改变而发生化学位移。
溶剂峰化学位移又称为线性化学位移(Linear Chemical Shift),按照物质形成的三维结构改变而改变的定义,可以将溶剂峰化学位移定义为有序变化化学位移。
因此,溶剂峰化学位移是一种有比较整齐的曲线,在某一原子化学位置上有一峰值,然后随着溶剂不同改变而发生有规律的化学位移。
溶剂峰化学位移的有效性及应用受到诸多因素的影响,包括基团原子的结构、形位和互变异构体配位以及尺寸变化换位的影响,对基团的位置极化的影响,和溶剂的离子手性的影响等。
特别是当基团量子效应出现时,溶剂峰化学位移就会发生较大的变化,包括轴向和同种溶剂上不同峰值等。
比较典型的溶剂峰化学位移情形有水平型、P-键、K抛物线型及曲线型等。
溶剂峰化学位移在聚合物相互作用研究、合成化学及活性立体化学等方面都有着广泛的应用。
当同一种原子组成不同**、优势体系或歪斜取向时,其随溶剂改变而发生的化学位移可以清楚的反映出来。
此外,溶剂峰化学位移可以用于判断多种结构的立体位置,或用来分析整个基团结构的改变。
因此,溶剂峰化学位移在核磁共振实验中是一种常用而有效的方法,为理解物质结构及相互作用提供了重要的技术保障。
核磁共振1H化学位移图表
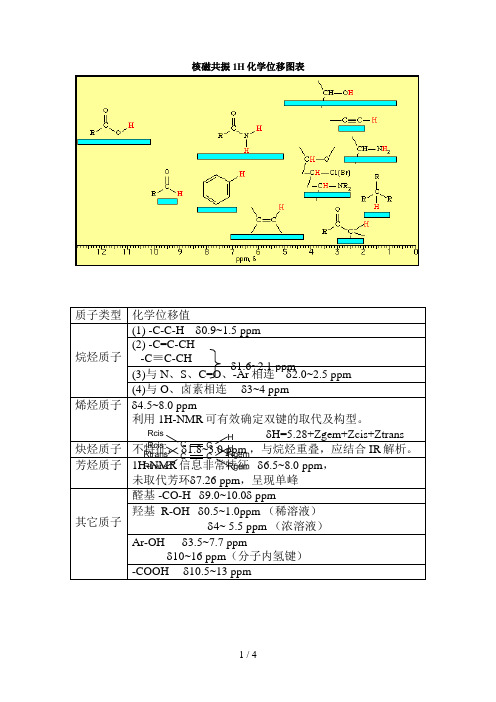
质子类型 化学位移值
烷烃质子 烯烃质子
(1) -C-C-H 0.9~1.5 ppm
(2) -C=C-CH (3-)C与≡NC、-CSH、C=O、-1A.r6相 ~2连.1 ppm2.0~2.5 ppm (4)与 O、卤素相连 1.63~~24.1ppm 4.5~8.0 ppm 利用 1H-NMR 可有效2.1确定双键的取代及构型。
CH3(t) 1.26 1.20 1.17 0.92 1.20 1.24 1.24 甲乙酮
CH3CO 2.14 2.07 2.07 1.58 2.06 2.12 2.19
CH2(q) 2.46 2.45 2.43 1.81 2.43 2.50 3.18
CH3(t) 1.06 0.96 0.91 0.85 0.96 1.01 1.26 乙二醇 —
CH3 2.88 2.78 2.73 1.86 2.77 2.86 2.85 二甲基亚砜— 2.62 2.52 2.54 1.68 2.50 2.65 2.71 二氧杂环 — 3.71 3.59 3.57 3.35 3.60 3.66 3.75 乙醇
CH3(t) 1.25 1.12 1.06 0.96 1.12 1.19 1.17
CH3(t) 1.21 1.11 1.09 1.11 1.12 1.18 1.17
CH2(q) 3.48 3.41 3.38 3.26 3.42 3.49 3.56 二甲基甲酰胺
CH 8.02 7.96 7.95 7.63 7.92 7.79 7.92
CH3 2.96 2.94 2.89 2.36 2.89 2.99 3.01
2/4
OH — — 4.19 1.55 2.18 — — 叔丁基甲醚
不同溶剂打核磁化学位移

不同溶剂打核磁化学位移核磁共振(NMR)是一种常用的谱学技术,用于研究化合物的分子结构和化学环境。
在NMR谱图中,化学位移是一个重要的参数,用来确定原子核的电子密度分布和化学环境。
不同溶剂对于核磁化学位移有着显著的影响,本文将一步一步回答“不同溶剂打核磁化学位移”。
第一步:了解核磁化学位移核磁化学位移指的是原子核相对于参考化合物的化学位移差异。
它以部位频率的偏离值(以部位频率为单位)表示,通常以ppm(parts per million)为单位进行表示。
核磁化学位移可以被许多因素影响,包括化学环境、分子结构和溶剂效应等。
第二步:溶剂效应的基本概念溶剂效应是指溶剂对分子结构和性质的影响。
在核磁共振谱图中,溶剂效应可能会导致化学位移的偏移。
溶剂分子与被溶解的化合物之间的相互作用可能会改变化合物的电子密度分布,从而影响核磁共振信号的位置和强度。
第三步:溶剂对核磁化学位移的影响不同溶剂对核磁化学位移的影响程度有所不同。
一般来说,溶剂分子中的质子会与待测化合物的质子之间发生相互作用,这种相互作用会导致待测化合物的核磁共振信号发生偏移。
这种偏移被称为溶剂效应。
在常见的溶剂中,二甲基亚砜(DMSO)、二氯甲烷(CDCl3)、乙酸(D6-acetone)、乙腈(D6-acetonitrile)和甲醇(CD3OH)等都被广泛应用于核磁共振实验中。
它们对于特定的化合物可能会产生不同的溶剂效应。
第四步:解释不同溶剂对核磁化学位移的影响解释不同溶剂对核磁化学位移的影响可以从两个方面进行讨论:1. 溶剂对电子密度的影响:溶剂分子中的质子会与待测化合物的质子发生相互作用,这种相互作用可能会导致化合物的电子密度发生变化。
这种变化可以通过溶剂效应的大小来定量表征。
溶剂分子的电子云密度会逐渐扩展到待测化合物的分子中,导致核磁共振信号的位置发生偏移。
2. 溶剂的极性和溶剂骨架的影响:溶剂的极性和溶剂骨架的特性可能会对化合物的电子密度分布产生影响,从而改变核磁共振信号的位置。
化学位移的单位

化学位移的单位核磁共振中,化学位移本身的单位并不是ppm,而其单位是Hz,之所以单位为ppm,是因为我们常说的化学位移指的是化学相对位移。
打个比方,当使用200MHz的NMR时,某个位移值为200Hz,这时就采用相对位移,用200Hz去除以200MHz,得到的是百万分之一,也就是1ppm;之所以这么表示是因为,位移值会随着机器的不同而改变,例如刚才的例子,在400MHz的NMR下,位移值是400Hz,只是相对位移不变,仍然是1ppm。
化学位移的公式表示:现采用相对数值表示法,即选用一个标准物质,以该标准物的共振吸收峰所处位置为零点,其它吸收峰的化学位移值根据这些吸收峰的位置与零点的距离来确定。
化学位移值普遍采用无量纲的δ值表示,其定义为:化学位移取决于核外电子云密度,因此影响电子云密度的各种因素都对化学位移有影响,影响最大的是电负性和各向异性效应。
1. 电负性电负性大的原子(或基团)吸电子能力强,降低了氢核外围的电子云密度,屏蔽效应也就随之降低,其共振吸收峰移向低场,化学位移会变大;反之,给电子基团可增加氢核外围的电子云密度,共振吸收峰移向高场,化学位移会变小。
2. 各向异性效应当分子中的某些基团的电子云排布不呈球形对称时,它对邻近的1H核产生一个各向异性的磁场,从而使某些空间位置上的核受屏蔽,而另一些空间位置上的核去屏蔽,这一现象称为各向异性效应(anisotropic effect)。
各向异性效应是由于成键电子的电子云分布不均匀导致在外磁场中所产生的感应磁场的不均匀所引起的,如苯环上质子的化学位移移向低场,δ在7左右。
3. 氢键氢键对羟基质子化学位移的影响与氢键的强弱及氢键的电子给予体的性质有关,在大多数情况下,氢键产生去屏蔽效应,使1H的δ值移向低场。
4. 溶剂效应有时同一种样品使用不同的溶剂也会使化学位移值发生变化,这称为溶剂效应。
活泼氢的溶剂效应比较明显。
能引起溶剂效应的因素很多,如N,N-二甲基甲酰胺在CDCl3中测定时,δαH>δβH,而在被测物中加入适量苯溶剂后可使δαH<δβH, 这是因为苯能与之形成复合物,而使两种氢处于不同的屏蔽区所致。
核磁氢谱溶剂峰化学位移表__解释说明

核磁氢谱溶剂峰化学位移表解释说明1. 引言1.1 概述核磁氢谱溶剂峰化学位移表是化学分析中非常重要的工具之一。
在核磁共振(NMR)技术中,溶剂峰是指由于溶剂中特定原子核的共振信号所引起的信号峰。
这些溶剂峰可以提供有关样品分子结构和化学环境的宝贵信息。
本篇文章将详细介绍核磁氢谱溶剂峰化学位移表的概念、意义以及构建方法,并解释如何使用该表进行核磁氢谱数据分析和解读。
1.2 文章结构本文将分为五个主要部分进行讨论。
首先,在引言部分,我们会对本文作出概述,并介绍文章内容和结构。
然后,我们将在第二部分介绍核磁氢谱溶剂峰的基本概念以及其在化学位移中的意义。
接着,我们将在第三部分详细探讨建立核磁氢谱溶剂峰化学位移表的方法。
在第四部分,我们将通过实际应用案例来说明如何分析和解读核磁氢谱溶剂峰化学位移表。
我们将介绍应用案例的背景,并阐述如何使用化学位移表来解读样品核磁氢谱数据。
最后,我们会讨论实际应用中可能遇到的挑战,并提出相应的解决方案。
最后,在结论与展望部分,我们将总结本文的研究成果,并对未来相关研究方向进行展望。
1.3 目的本文旨在全面介绍核磁氢谱溶剂峰化学位移表及其分析和解释方法,以帮助读者更好地理解和运用这一重要工具。
通过对该表的深入了解,读者可以准确地分析和解读核磁氢谱数据,并在实际应用中有效利用溶剂峰化学位移信息进行样品结构和环境的推测。
2. 核磁氢谱溶剂峰化学位移表:2.1 核磁氢谱概述:核磁共振(NMR)是一种重要的分析技术,广泛应用于化学和生物学领域。
核磁氢谱是其中一种常见的NMR实验,用于确定分子中氢原子的化学环境和相互作用。
在核磁氢谱图中,峰表示不同化学位移的氢原子信号。
2.2 溶剂峰化学位移的意义:在进行核磁氢谱测定时,需要选择一个特定的溶剂作为溶剂系统的参考标准。
这个溶剂在谱图中会产生一个固定位置的峰,称为溶剂峰。
通过与溶剂峰对比,可以精确地确定其他化合物中氢原子信号的化学位移。
溶剂峰化学位移表是记录各种常见有机溶剂在核磁共振实验中对应峰位置(通常以ppm表示)的表格。
核磁溶剂峰列表

NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities Hugo E.Gottlieb,*Vadim Kotlyar,andAbraham Nudelman*Department of Chemistry,Bar-Ilan University,Ramat-Gan52900,IsraelReceived June27,1997In the course of the routine use of NMR as an aid for organic chemistry,a day-to-day problem is the identifica-tion of signals deriving from common contaminants (water,solvents,stabilizers,oils)in less-than-analyti-cally-pure samples.This data may be available in the literature,but the time involved in searching for it may be considerable.Another issue is the concentration dependence of chemical shifts(especially1H);results obtained two or three decades ago usually refer to much more concentrated samples,and run at lower magnetic fields,than today’s practice.We therefore decided to collect1H and13C chemical shifts of what are,in our experience,the most popular “extra peaks”in a variety of commonly used NMR solvents,in the hope that this will be of assistance to the practicing chemist.Experimental SectionNMR spectra were taken in a Bruker DPX-300instrument (300.1and75.5MHz for1H and13C,respectively).Unless otherwise indicated,all were run at room temperature(24(1°C).For the experiments in the last section of this paper,probe temperatures were measured with a calibrated Eurotherm840/T digital thermometer,connected to a thermocouple which was introduced into an NMR tube filled with mineral oil to ap-proximately the same level as a typical sample.At each temperature,the D2O samples were left to equilibrate for at least 10min before the data were collected.In order to avoid having to obtain hundreds of spectra,we prepared seven stock solutions containing approximately equal amounts of several of our entries,chosen in such a way as to prevent intermolecular interactions and possible ambiguities in assignment.Solution1:acetone,tert-butyl methyl ether,di-methylformamide,ethanol,toluene.Solution2:benzene,di-methyl sulfoxide,ethyl acetate,methanol.Solution3:acetic acid,chloroform,diethyl ether,2-propanol,tetrahydrofuran. Solution4:acetonitrile,dichloromethane,dioxane,n-hexane, HMPA.Solution5:1,2-dichloroethane,ethyl methyl ketone, n-pentane,pyridine.Solution6:tert-butyl alcohol,BHT,cyclo-hexane,1,2-dimethoxyethane,nitromethane,silicone grease, triethylamine.Solution7:diglyme,dimethylacetamide,ethyl-ene glycol,“grease”(engine oil).For D2O.Solution1:acetone, tert-butyl methyl ether,dimethylformamide,ethanol,2-propanol. Solution2:dimethyl sulfoxide,ethyl acetate,ethylene glycol, methanol.Solution3:acetonitrile,diglyme,dioxane,HMPA, pyridine.Solution4:1,2-dimethoxyethane,dimethylacetamide, ethyl methyl ketone,triethylamine.Solution5:acetic acid,tert-butyl alcohol,diethyl ether,tetrahydrofuran.In D2O and CD3OD nitromethane was run separately,as the protons exchanged with deuterium in presence of triethylamine.ResultsProton Spectra(Table1).A sample of0.6mL of the solvent,containing1µL of TMS,1was first run on its own.From this spectrum we determined the chemical shifts of the solvent residual peak2and the water peak. It should be noted that the latter is quite temperature-dependent(vide infra).Also,any potential hydrogen-bond acceptor will tend to shift the water signal down-field;this is particularly true for nonpolar solvents.In contrast,in e.g.DMSO the water is already strongly hydrogen-bonded to the solvent,and solutes have only a negligible effect on its chemical shift.This is also true for D2O;the chemical shift of the residual HDO is very temperature-dependent(vide infra)but,maybe counter-intuitively,remarkably solute(and pH)independent. We then added3µL of one of our stock solutions to the NMR tube.The chemical shifts were read and are presented in Table 1.Except where indicated,the coupling constants,and therefore the peak shapes,are essentially solvent-independent and are presented only once.For D2O as a solvent,the accepted reference peak(δ)0)is the methyl signal of the sodium salt of3-(trimeth-ylsilyl)propanesulfonic acid;one crystal of this was added to each NMR tube.This material has several disadvan-tages,however:it is not volatile,so it cannot be readily eliminated if the sample has to be recovered.In addition, unless one purchases it in the relatively expensive deuterated form,it adds three more signals to the spectrum(methylenes1,2,and3appear at2.91,1.76, and0.63ppm,respectively).We suggest that the re-sidual HDO peak be used as a secondary reference;we find that if the effects of temperature are taken into account(vide infra),this is very reproducible.For D2O, we used a different set of stock solutions,since many of the less polar substrates are not significantly water-soluble(see Table1).We also ran sodium acetate and sodium formate(chemical shifts: 1.90and8.44ppm, respectively).Carbon Spectra(Table2).To each tube,50µL of the stock solution and3µL of TMS1were added.The solvent chemical shifts3were obtained from the spectra containing the solutes,and the ranges of chemical shifts(1)For recommendations on the publication of NMR data,see: IUPAC Commission on Molecular Structure and Spectroscopy.Pure Appl.Chem.1972,29,627;1976,45,217.(2)I.e.,the signal of the proton for the isotopomer with one less deuterium than the perdeuterated material,e.g.,C H Cl3in CDCl3or C6D5H in C6D6.Except for CHCl3,the splitting due to J HD is typically observed(to a good approximation,it is1/6.5of the value of the corresponding J HH).For CHD2groups(deuterated acetone,DMSO, acetonitrile),this signal is a1:2:3:2:1quintet with a splitting of ca.2 Hz.(3)In contrast to what was said in note2,in the13C spectra the solvent signal is due to the perdeuterated isotopomer,and the one-bond couplings to deuterium are always observable(ca.20-30Hz). Figure1.Chemical shift of H DO as a function of tempera-ture..Chem.1997,62,7512-7515S0022-3263(97)01176-6CCC:$14.00©1997American Chemical Societyshow their degree of variability.Occasionally,in order to distinguish between peaks whose assignment was ambiguous,a further1-2µL of a specific substrate were added and the spectra run again.Table1.1H NMR Dataproton mult CDCl3(CD3)2CO(CD3)2SO C6D6CD3CN CD3OD D2O solvent residual peak7.26 2.05 2.507.16 1.94 3.31 4.79 H2O s 1.56 2.84a 3.33a0.40 2.13 4.87acetic acid CH3s 2.10 1.96 1.91 1.55 1.96 1.99 2.08 acetone CH3s 2.17 2.09 2.09 1.55 2.08 2.15 2.22 acetonitrile CH3s 2.10 2.05 2.07 1.55 1.96 2.03 2.06 benzene CH s7.367.367.377.157.377.33tert-butyl alcohol CH3s 1.28 1.18 1.11 1.05 1.16 1.40 1.24 OH c s 4.19 1.55 2.18tert-butyl methyl ether CCH3s 1.19 1.13 1.11 1.07 1.14 1.15 1.21 OCH3s 3.22 3.13 3.08 3.04 3.13 3.20 3.22 BHT b ArH s 6.98 6.96 6.877.05 6.97 6.92OH c s 5.01 6.65 4.79 5.20ArCH3s 2.27 2.22 2.18 2.24 2.22 2.21ArC(CH3)3s 1.43 1.41 1.36 1.38 1.39 1.40chloroform CH s7.268.028.32 6.157.587.90 cyclohexane CH2s 1.43 1.43 1.40 1.40 1.44 1.451,2-dichloroethane CH2s 3.73 3.87 3.90 2.90 3.81 3.78 dichloromethane CH2s 5.30 5.63 5.76 4.27 5.44 5.49diethyl ether CH3t,7 1.21 1.11 1.09 1.11 1.12 1.18 1.17 CH2q,7 3.48 3.41 3.38 3.26 3.42 3.49 3.56 diglyme CH2m 3.65 3.56 3.51 3.46 3.53 3.61 3.67 CH2m 3.57 3.47 3.38 3.34 3.45 3.58 3.61OCH3s 3.39 3.28 3.24 3.11 3.29 3.35 3.37 1,2-dimethoxyethane CH3s 3.40 3.28 3.24 3.12 3.28 3.35 3.37 CH2s 3.55 3.46 3.43 3.33 3.45 3.52 3.60 dimethylacetamide CH3CO s 2.09 1.97 1.96 1.60 1.97 2.07 2.08 NCH3s 3.02 3.00 2.94 2.57 2.96 3.31 3.06NCH3s 2.94 2.83 2.78 2.05 2.83 2.92 2.90 dimethylformamide CH s8.027.967.957.637.927.977.92 CH3s 2.96 2.94 2.89 2.36 2.89 2.99 3.01CH3s 2.88 2.78 2.73 1.86 2.77 2.86 2.85 dimethyl sulfoxide CH3s 2.62 2.52 2.54 1.68 2.50 2.65 2.71 dioxane CH2s 3.71 3.59 3.57 3.35 3.60 3.66 3.75 ethanol CH3t,7 1.25 1.12 1.060.96 1.12 1.19 1.17 CH2q,7d 3.72 3.57 3.44 3.34 3.54 3.60 3.65OH s c,d 1.32 3.39 4.63 2.47ethyl acetate CH3CO s 2.05 1.97 1.99 1.65 1.97 2.01 2.07C H2CH3q,7 4.12 4.05 4.03 3.89 4.06 4.09 4.14CH2C H3t,7 1.26 1.20 1.170.92 1.20 1.24 1.24 ethyl methyl ketone CH3CO s 2.14 2.07 2.07 1.58 2.06 2.12 2.19C H2CH3q,7 2.46 2.45 2.43 1.81 2.43 2.50 3.18CH2C H3t,7 1.060.960.910.850.96 1.01 1.26 ethylene glycol CH s e 3.76 3.28 3.34 3.41 3.51 3.59 3.65“grease”f CH3m0.860.870.920.860.88CH2br s 1.26 1.29 1.36 1.27 1.29n-hexane CH3t0.880.880.860.890.890.90CH2m 1.26 1.28 1.25 1.24 1.28 1.29HMPA g CH3d,9.5 2.65 2.59 2.53 2.40 2.57 2.64 2.61 methanol CH3s h 3.49 3.31 3.16 3.07 3.28 3.34 3.34 OH s c,h 1.09 3.12 4.01 2.16nitromethane CH3s 4.33 4.43 4.42 2.94 4.31 4.34 4.40 n-pentane CH3t,70.880.880.860.870.890.90CH2m 1.27 1.27 1.27 1.23 1.29 1.292-propanol CH3d,6 1.22 1.10 1.040.95 1.09 1.50 1.17 CH sep,6 4.04 3.90 3.78 3.67 3.87 3.92 4.02 pyridine CH(2)m8.628.588.588.538.578.538.52 CH(3)m7.297.357.39 6.667.337.447.45CH(4)m7.687.767.79 6.987.737.857.87 silicone grease i CH3s0.070.130.290.080.10 tetrahydrofuran CH2m 1.85 1.79 1.76 1.40 1.80 1.87 1.88 CH2O m 3.76 3.63 3.60 3.57 3.64 3.71 3.74 toluene CH3s 2.36 2.32 2.30 2.11 2.33 2.32CH(o/p)m7.177.1-7.27.187.027.1-7.37.16CH(m)m7.257.1-7.27.257.137.1-7.37.16 triethylamine CH3t,7 1.030.960.930.960.96 1.050.99 CH2q,7 2.53 2.45 2.43 2.40 2.45 2.58 2.57a In these solvents the intermolecular rate of exchange is slow enough that a peak due to HDO is usually also observed;it appears at2.81and3.30ppm in acetone and DMSO,respectively.In the former solvent,it is often seen as a1:1:1triplet,with2J H,D)1Hz. b2,6-Dimethyl-4-tert-butylphenol.c The signals from exchangeable protons were not always identified.d In some cases(see note a),the coupling interaction between the CH2and the OH protons may be observed(J)5Hz).e In CD3CN,the OH proton was seen as a multiplet atδ2.69,and extra coupling was also apparent on the methylene peak.f Long-chain,linear aliphatic hydrocarbons.Their solubility in DMSO was too low to give visible peaks.g Hexamethylphosphoramide.h In some cases(see notes a,d),the coupling interaction between the CH3and the OH protons may be observed(J)5.5Hz).i Poly(dimethylsiloxane).Its solubility in DMSO was too low to give visible peaks.Notes .Chem.,Vol.62,No.21,19977513.Chem.,Vol.62,No.21,1997NotesTable2.13C NMR Data aCDCl3(CD3)2CO(CD3)2SO C6D6CD3CN CD3OD D2O solvent signals77.16(0.0629.84(0.0139.52(0.06128.06(0.02 1.32(0.0249.00(0.01206.26(0.13118.26(0.02acetic acid CO175.99172.31171.93175.82173.21175.11177.21 CH320.8120.5120.9520.3720.7320.5621.03 acetone CO207.07205.87206.31204.43207.43209.67215.94 CH330.9230.6030.5630.1430.9130.6730.89 acetonitrile CN116.43117.60117.91116.02118.26118.06119.68 CH3 1.89 1.12 1.030.20 1.790.85 1.47 benzene CH128.37129.15128.30128.62129.32129.34tert-butyl alcohol C69.1568.1366.8868.1968.7469.4070.36 CH331.2530.7230.3830.4730.6830.9130.29 tert-butyl methyl ether OCH349.4549.3548.7049.1949.5249.6649.37 C72.8772.8172.0472.4073.1774.3275.62C C H326.9927.2426.7927.0927.2827.2226.60 BHT C(1)151.55152.51151.47152.05152.42152.85C(2)135.87138.19139.12136.08138.13139.09CH(3)125.55129.05127.97128.52129.61129.49C(4)128.27126.03124.85125.83126.38126.11CH3Ar21.2021.3120.9721.4021.2321.38C H3C30.3331.6131.2531.3431.5031.15C34.2535.0034.3334.3535.0535.36chloroform CH77.3679.1979.1677.7979.1779.44cyclohexane CH226.9427.5126.3327.2327.6327.961,2-dichloroethane CH243.5045.2545.0243.5945.5445.11 dichloromethane CH253.5254.9554.8453.4655.3254.78diethyl ether CH315.2015.7815.1215.4615.6315.4614.77 CH265.9166.1262.0565.9466.3266.8866.42 diglyme CH359.0158.7757.9858.6658.9059.0658.67 CH270.5171.0369.5470.8770.9971.3370.05CH271.9072.6371.2572.3572.6372.9271.63 1,2-dimethoxyethane CH359.0858.4558.0158.6858.8959.0658.67 CH271.8472.4717.0772.2172.4772.7271.49 dimethylacetamide CH321.5321.5121.2921.1621.7621.3221.09 CO171.07170.61169.54169.95171.31173.32174.57NCH335.2834.8937.3834.6735.1735.5035.03NCH338.1337.9234.4237.0338.2638.4338.76 dimethylformamide CH162.62162.79162.29162.13163.31164.73165.53 CH336.5036.1535.7335.2536.5736.8937.54CH331.4531.0330.7330.7231.3231.6132.03 dimethyl sulfoxide CH340.7641.2340.4540.0341.3140.4539.39 dioxane CH267.1467.6066.3667.1667.7268.1167.19 ethanol CH318.4118.8918.5118.7218.8018.4017.47 CH258.2857.7256.0757.8657.9658.2658.05 ethyl acetate C H3CO21.0420.8320.6820.5621.1620.8821.15 CO171.36170.96170.31170.44171.68172.89175.26CH260.4960.5659.7460.2160.9861.5062.32CH314.1914.5014.4014.1914.5414.4913.92 ethyl methyl ketone C H3CO29.4929.3029.2628.5629.6029.3929.49 CO209.56208.30208.72206.55209.88212.16218.43C H2CH336.8936.7535.8336.3637.0937.3437.27CH2C H37.868.037.617.918.148.097.87 ethylene glycol CH263.7964.2662.7664.3464.2264.3063.17“grease”CH229.7630.7329.2030.2130.8631.29n-hexane CH314.1414.3413.8814.3214.4314.45CH2(2)22.7023.2822.0523.0423.4023.68CH2(3)31.6432.3030.9531.9632.3632.73HMPA b CH336.8737.0436.4236.8837.1037.0036.46 methanol CH350.4149.7748.5949.9749.9049.8649.50c nitromethane CH362.5063.2163.2861.1663.6663.0863.22 n-pentane CH314.0814.2913.2814.2514.3714.39CH2(2)22.3822.9821.7022.7223.0823.38CH2(3)34.1634.8333.4834.4534.8935.302-propanol CH325.1425.6725.4325.1825.5525.2724.38 CH64.5063.8564.9264.2364.3064.7164.88 pyridine CH(2)149.90150.67149.58150.27150.76150.07149.18 CH(3)123.75124.57123.84123.58127.76125.53125.12CH(4)135.96136.56136.05135.28136.89138.35138.27 silicone grease CH3 1.04 1.40 1.38 2.10 tetrahydrofuran CH225.6226.1525.1425.7226.2726.4825.67 CH2O67.9768.0767.0367.8068.3368.8368.68 toluene CH321.4621.4620.9921.1021.5021.50C(i)137.89138.48137.35137.91138.90138.85CH(o)129.07129.76128.88129.33129.94129.91CH(m)128.26129.03128.18128.56129.23129.20CH(p)125.33126.12125.29125.68126.28126.29triethylamine CH311.6112.4911.7412.3512.3811.099.07 CH246.2547.0745.7446.7747.1046.9647.19a See footnotes for Table1.b2J PC)3Hz.c Reference material;see text.For D2O solutions there is no accepted reference for carbon chemical shifts.We suggest the addition of a drop of methanol,and the position of its signal to be defined as49.50ppm;on this basis,the entries in Table2were recorded.The chemical shifts thus obtained are,on the whole,very similar to those for the other solvents. Alternatively,we suggest the use of dioxane when the methanol peak is expected to fall in a crowded area of the spectrum.We also report the chemical shifts of sodium formate(171.67ppm),sodium acetate(182.02and 23.97ppm),sodium carbonate(168.88ppm),sodium bicarbonate(161.08ppm),and sodium3-(trimethylsilyl)-propanesulfonate[54.90,19.66,15.56(methylenes1,2, and3,respectively),and-2.04ppm(methyls)],in D2O. Temperature Dependence of HDO Chemical Shifts.We recorded the1H spectrum of a sample of D2O, containing a crystal of sodium3-(trimethylsilyl)propane-sulfonate as reference,as a function of temperature.The data are shown in Figure1.The solid line connecting the experimental points corresponds to the equation which reproduces the measured values to better than1 ppb.For the0-50o C range,the simplergives values correct to10ppb.For both equations,T is the temperature in°C.Acknowledgment.Generous support for this work by the Minerva Foundation and the Otto Mayerhoff Center for the Study of Drug-Receptor Interactions at Bar-Ilan University is gratefully acknowledged.JO971176Vδ)5.060-0.0122T+(2.11×10-5)T2(1)δ)5.051-0.0111T(2)Notes .Chem.,Vol.62,No.21,19977515。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
测试核磁的样品一般要求比较纯,并且能够溶解在氘代试剂中,这样才能测得高分辨率的图谱。
为不干扰谱图,所用溶剂分子中的氢都应被氘取代,但难免有氢的残余(1%左右),这样就会产生溶剂峰;除了残存的质子峰外,溶剂中有时会有微量的H2O而产生水峰,而且这个H2O峰的位置也会因溶剂的不同而不同;另外,在样品(或制备过程)中,也难免会残留一些杂质,在图谱上就会有杂质峰,应注意识别。
常用氘代溶剂和杂质峰在1H谱中的化学位移单位:ppm
溶剂—CDCl3 (CD3)2CO (CD3)2SO C6D6 CD3CN CD3OH D2O
溶剂峰—7.26 2.05 2.49 7.16 1.94 3.31 4.80
水峰— 1.56 2.84 3.33 0.40 2.13 4.87 —
乙酸— 2.10 1.96 1.91 1.55 1.96 1.99 2.08
丙酮— 2.17 2.09 2.09 1.55 2.08 2.15 2.22
乙腈— 2.10 2.05 2.07 1.55 1.96 2.03 2.06
苯—7.36 7.36 7.37 7.15 7.37 7.33 —
叔丁醇CH3 1.28 1.18 1.11 1.05 1.16 1.40 1.24 OH —— 4.19 1.55 2.18 ——
叔丁基甲醚
CCH3 1.19 1.13 1.11 1.07 1.14 1.15 1.21
OCH3 3.22 3.13 3.08 3.04 3.13 3.20 3.22
氯仿—7.26 8.02 8.32 6.15 7.58 7.90 —
环己烷— 1.43 1.43 1.40 1.40 1.44 1.45 —
1,2-二氯甲烷 3.73 3.87 3.90 2.90 3.81 3.78 —
二氯甲烷— 5.30 5.63 5.76 4.27 5.44 5.49 —
乙醚 CH3(t) 1.21 1.11 1.09 1.11 1.12 1.18 1.17 CH2(q) 3.48 3.41 3.38 3.26 3.42 3.49 3.56
二甲基甲酰胺
CH 8.02 7.96 7.95 7.63 7.92 7.79 7.92
CH3 2.96 2.94 2.89 2.36 2.89 2.99 3.01
CH3 2.88 2.78 2.73 1.86 2.77 2.86 2.85
二甲基亚砜— 2.62 2.52 2.54 1.68 2.50 2.65 2.71
二氧杂环— 3.71 3.59 3.57 3.35 3.60 3.66 3.75
乙醇 CH3(t) 1.25 1.12 1.06 0.96 1.12 1.19 1.17 CH2(q) 3.72 3.57 3.44 3.34 3.54 3.60 3.65
OH(s) 1.32 3.39 3.63 — 2.47 ——
乙酸乙酯CH3CO 2.05 1.97 1.99 1.65 1.97 2.01 2.07 OCH2(q) 4.12 4.05 4.03 3.89 4.06 4.09 4.14
CH3(t) 1.26 1.20 1.17 0.92 1.20 1.24 1.24 甲乙酮CH3CO 2.14 2.07 2.07 1.58 2.06 2.12 2.19 CH2(q) 2.46 2.45 2.43 1.81 2.43 2.50 3.18
CH3(t) 1.06 0.96 0.91 0.85 0.96 1.01 1.26 乙二醇— 3.76 3.28 3.34 3.41 3.51 3.59 3.65
润滑脂 CH3(m) 0.86 0.87 —0.92 0.86 0.88 —CH2(br) 1.26 1.29 — 1.36 1.27 1.29 —
正己烷CH3(t) 0.88 0.88 0.86 0.89 0.89 0.90 —CH2 (m) 1.26 1.28 1.25 1.24 1.28 1.29 —
甲醇CH3 3.49 3.31 3.16 3.07 3.28 3.34 3.34 OH 1.09 3.12 4.01 2.16 ——
正戊烷 CH3(t) 0.88 0.88 0.86 0.87 0.89 0.90 —CH2(m) 1.27 1.27 1.27 1.23 1.29 1.29 —
异丙醇CH3(d) 1.22 1.10 1.04 0.95 1.09 1.50 1.17 CH 4.04 3.90 3.78 3.67 3.87 3.92 4.02 硅脂—0.07 0.13 —0.29 0.08 0.10 —
四氢呋喃 CH2 1.85 1.79 1.76 1.40 1.80 1.87 1.88 CH2O 3.76 3.63 3.60 3.57 3.64 3.71 3.74 甲苯 CH3 2.36 2.32 2.30 2.11 2.33 2.32 —CH(o/p)7.17 7.20 7.18 7.02 7.30 7.16 —
CH(m) 7.25 7.20 7.25 7.13 7.30 7.16 —
三乙基胺 CH3 1.03 0.96 0.93 0.96 0.96 1.05 0.99 CH2 2.53 2.45 2.43 2.40 2.45 2.58 2.57 石油醚—0.5-1.5 0.6-1.9 —————。