注射用无菌粉末装量差异检查法标准操作规程
注射剂的常规检查项目与方法

可复测20支,均不得检出。
灯检法仪器
注射剂的常规检查项目与方法
四、不溶性微粒
可见异物检查,只能检出50um以上微粒。 在可见异物检查符合规定后,检查溶液型静脉用 注射液中不溶性微粒的大小及数量。 检查方法:显微计数法
光阻法。
注射剂的常规检查项目与方法
五、无菌、细菌内毒素或热源
(一)无菌检查 1. 100级洁净区进行,全过程应严格遵守无菌操作,防止 微生物污染。 2. 检查方法:
二、装量差异
注射用无菌粉末检查装量差异
如有1瓶不符合规定,应另取10瓶复试,应符合 规定。
注射剂的常规检查项目与方法
三、可见异物
(一)定义
指存在于注射液、滴眼液中,在规定条件下目视可以
观测到的不溶性物质,其粒径或长度通常大于50µm。
(二)检查方法
1. 灯检法
2. 光散射法
(三)判定结果
取20支,若1支不合格,
制作人|
注射剂的常规检查项目与 方法
注射剂的常规检查项目与方法
一、装量
取样量:≤2ml,5支
将内容物分别用
>2ml至50ml,3支
相应体积的干燥 注射器及注射针
测定方法:体积测量
头抽尽,然后注
判定标准:每支装量均不得少于ຫໍສະໝຸດ 示量 入标化的量具内 ,在室温下检视
>50ml者,照“最低装量检查法”。
注射剂的常规检查项目与方法
(1)直接接种法 (2)薄膜过滤法
注射剂的常规检查项目与方法
五、无菌、细菌内毒素或热源
(二)细菌内毒素
细菌细胞壁的组分,由脂多糖组成。
细菌内毒素检查原理:鲎试剂与细菌内毒素的凝集反应。
方法: 1. 凝胶法
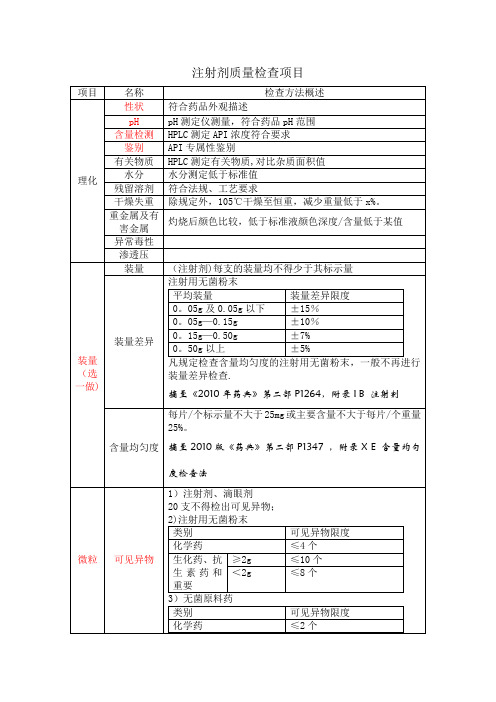
注射剂质量检查
每 1ml:≥10um 微粒低于 12 粒;≥ 25um 微粒低于 3 粒
≥10ml 微粒低于 6000 粒; ≥25um 微粒低于 600 粒子
>100ml <100ml
每 1ml:≥10um 微粒低于 12 粒; ≥25um 微粒低于 2 粒
≥10ml 微粒低于 3000 粒; ≥25um 微粒低于 300 粒子
灼烧后颜色比较,低于标准液颜色深度/含量低于某值
(注射剂)每支的装量均不得少于其标示量
注射用无菌粉末
平均装量
装量差异限度
0。05g 及 0.05g 以下 ±15%
0。05g—0.15g
±10%
0。15g—0.50g
±7%
0。50g 以上
±5%
凡规定检查含量均匀度的注射用无菌粉末,一般不再进行
装量差异检查.
注射剂质量检查项目
项目 理化
装量 (选 一做)
名称 性状 pH 含量检测 鉴别 有关物质 水分 残留溶剂 干燥失重 重金属及有 害金属 异常毒性 渗透压 装量
装量差异
检查方法概述 符合药品外观描述 pH 测定仪测量,符合药品 pH 范围 HPLC 测定 API 浓度符合要求 API 专属性鉴别 HPLC 测定有关物质,对比杂质面积值 水分测定低于标准值 符合法规、工艺要求 除规定外,105℃干燥至恒重,减少重量低于 x%。
无菌检查 供试品全部无菌
1)热原—家兔法;
标准:在初试的 3 只家兔中,体温升高均低于 0.6℃,并且
3 只家兔体温升高总和低于 1.3℃;或在复试的 5 只家兔中,
无菌
内毒素/热 原
体温升高 0.6℃或高于 0。6℃的家兔不超过 1 只,并且初 试、复试合并 8 只家兔的体温升高总和为 3.5℃或低于 3.5℃,均判定供试品的热原检查符合规定。
中药制剂常规检查技术—重(装)量差异检查法

根据标示装量,查表确定装量差异限度
丸 剂
根据装量差异限度,计算允许装量范围
进行结果判断:供试品装量超过装量差异限度允许范围的不多于2份,且均未超过限度1倍 ,判为合格
三、丸剂装量差异检查
4.应用实例
加味逍遥丸的装量差异检查
批号:2021412
标示装量:6g/袋
编号
1
2
3
4
5
6
7
8
9 10
装量 6.35 6.28 6.48 6.53 6.17 5.98 6.38 6.12 5.96 6.10
一、方法概述
1.定义 重量差异:药品本身的重量与药品标示重量或平均重量的偏差程度。
重量差异检查:以药物制剂的标示量或平均重量为基准,对药品重量的偏差程度进行检查, 从而评价制剂质量的均一性。
一、方法概述
2.检查对象
《中国药典》(2020年版)对固体中药制剂的重量差异检查做出明确规定:
根据药品的聚集状态,药品颗粒较集中的应进行重量差异检查,如大蜜丸、浓缩丸、小蜜丸、 滴丸剂、片剂、贴膏剂、栓剂、锭剂和膏药等。
二、胶囊剂装量差异检查
1.简述
胶囊剂在生产过程中由于包装工艺、设备和管理等原因,都会引起胶囊剂的装量差异。本项 检查的目的在于控制最小包装内药品重量的一致性,保证用药剂量准确。
注意:凡规定检查含量均匀度的胶囊剂,一般不再进行装量差异检查。 每粒胶囊的两次称量中,应注意编号顺序以及囊体和囊帽的对号,不得混淆。 在称量前后,均应仔细查对胶囊数。称量过程中应避免用手直接接触供试品。已取出的
三、丸剂重量差异检查
1.简述
由于丸剂的类型、包装及剂量规格的多样性,《中国药典》规定滴丸、糖丸、蜜丸、水蜜丸、 浓缩丸等应进行重量差异检查。 注意:包糖衣丸剂应在包衣前检查丸芯的重量差异,符合规定后方可包衣,包糖衣后不再检査 重量差异。
注射用无菌粉末装量差异检查法标准操作规程

注射用无菌粉末装量差异检查法标准操作规程目的:建立注射用无菌粉末装量差异检查法标准操作规程,控制各瓶间装量的一致性,以保证使用剂量的准确。
2.依据:《中华人民共和国药典》2000年版二部。
3. 范围:适用于橡皮塞铝盖玻瓶或安瓿装的注射用无菌粉末的装量差异检查。
4. 职责:QA检查员、QC检验员对本标准的实施负责。
5. 程序:5.1. 仪器与用具:分析天平感量1mg或0.1mg(适用于检查装量小于0.1g的粉针剂)5.2.检查法:5.2.1. 取供试品5瓶(支),除去铝盖和瓶签(若为纸标签,用水润湿后除去纸屑;若为直接在玻璃上印字标签,用适当有机溶媒擦除字迹),容器外壁用乙醇洗净,置干燥器内放置1-2小时,俟干燥后,分别编号,依次放于固定位置。
5.2.2.轻扣橡皮塞或安瓿颈,使其上附着的粉末全部落下,分别精密称定每瓶(支)的重量,开启容器(注意避免玻璃屑等异物落入容器中),倾出内容物,容器用水、乙醇洗净,依次放回原固定位置,在适当的条件下干燥后,再分别精密称定每一容器的重量,即可求出每1瓶(支)的装量和平均装量。
5.2.3.复试、初试中,如有1瓶的装量超过装量差异限度规定时,另取10瓶(支)按5.2.1.、5.2.2.项下复试。
5.3.记录与计算:5.3.1. 记录每次称量数据。
5.3.2.根据每瓶(支)的重量与其空瓶重之差,求算每瓶(支)内容物重量。
5.3.3.每瓶(支)内容物重量之和除以5(复试时除以10),即得平均装量( m ),保留3位有效数字。
5.3.4.按下表规定装量差异限度,求出允许装量范围(m±m×装量差异限度)。
注射用无菌粉末装量差异限度规定5.4.结果与判定:5.4.1. 每1瓶(支)中的装量与平均装量相比较,均末超过者,判为符合规定。
5.4.2.每1瓶(支)中的装量与平均装量相比较,超过1瓶者,判为不符合规定。
5.4.3.初试结果仅有1瓶(支)的装量超过允许装量范围时,另取10瓶(支)复试。
13实验十三 装量差异检查

实验十一装量差异检查一、实验目的1. 掌握注射用无菌粉末装量差异检查的步骤、结果和判断标准。
2. 熟练使用分析天平。
二、实验原理控制各瓶间装量的一致性,以保证使用剂量的准确。
《中国药典》规定,注射用无菌粉末平均装量0.30g以下时,需做装量差异检查。
注射用无菌粉末装量差异限度规定三、实验仪器与试剂1. 仪器:分析天平、2. 试剂:注射用青霉素钠(0.48g)四、实验内容1、注射用无菌粉末装量差异检查(1)实验内容及步骤取注射用青霉素钠(0.48g)5瓶,除去铝盖和瓶签(若为纸标签,用水润湿后除去纸屑;若为直接在玻璃上印字标签,用适当有机溶媒擦除字迹),容器外壁用乙醇洗净,置干燥器内放置1-2小时,等干燥后,分别编号,依次放于固定位置。
轻扣橡皮塞,使其上附着的粉末全部落下,分别精密称定每瓶的重量,开启容器(注意避免玻璃屑等异物落入容器中),倾出内容物,容器用水、乙醇洗净,依次放回原固定位置,在适当的条件下干燥后,再分别精密称定每一容器的重量,即可求出每1瓶(支)的装量和平均装量。
(2)记录与计算:1)记录每次称量数据。
2)计算a. 每瓶的内容物重量根据每瓶(支)的重量与其空瓶重之差,求算每瓶(支)内容物重量。
b. 平均装量每瓶(支)内容物重量之和除以5(复试时除以10),即得平均装量( m ),保留3位有效数字。
c. 按下表规定装量差异限度,求出允许装量范围(m±m×装量差异限度)。
d. 遇有超出允许装量范围并处于边缘者,应再与平均装量相比较,计算出该瓶装量差异的百分率,再根据上表规定的装量差异限度作为判定的依据(避免在计算允许装量范围时受数字修约的影响)。
3) 结果与判定:a. 每瓶中的装量均未超出允许装量范围(m±m×装量差异限度);或其每1瓶中的装量与平均装量相比较,均末超过者,判为符合规定。
b. 每1瓶中的装量与平均装量相比较,超过1瓶者,判为不符合规定。
注射剂装量差异的检查方法及标准

注射剂装量差异的检查方法及标准
检查方法:
1.选取一定数量的注射器进行检查。
2.准备一个注射器架,将注射器按照规定的容量放置在注射器
架上。
3.取出一支注射器,用注射器架固定在定量容器上,注意不要
造成外力影响。
4.将注射器慢慢按照规定体积进料,直至液位到指定位置。
5.用定量瓶或分析天平,测定进入注射器内的体积,求出与规
定体积的差异。
6.重复多次测量,然后计算出注射器装量差异的平均值和标准差。
标准:
1.药典要求不同体积的注射器的允许误差不同,常见的注射器
错误容量限度如下:0.1ml以下±0.02ml;0.2ml~2ml±0.03ml;5ml以上±0.05ml。
2.注射器装量的标准差应小于其误差容限的1/3。
3.检查结果应符合国家药典要求,否则产品不合格。
药品检验标准操作规程

5.7 测定时,除另有规定外,每个溶出杯只允许 投入供试品1片(粒、袋),不得多投,并应 注意投入杯底中心位置。
5.8 测定 紫外法测定时,应选择规定波长的 ±2nm,即5个波长的测定值中的最大值;如使 用吸收系数测定时,应在最大吸收波长测定吸 光度。
(五)水分测定 卡氏水分测定法 1.标化 平行测定三次,取其平均值,RSD≤1.0%
或采用煮沸、超声、抽滤等其他有效的除
气方法。如果溶出介质为缓冲液,当需 要调节pH值时,一般调节pH值至规定pH 值的±0.05之内。
将该品种项下所规定的溶出介质经脱气,并按规
定量置于溶出杯中,开启仪器预置温度,一般 应根据室温情况,可稍高于37℃,以使溶出杯 中溶出介质的温度保持在37±0.5℃,并应使用 0.1分度的温度计,逐一检查6个溶出杯中溶出 介质的温度,其间差异应在0.5℃之内。
空胶囊的干扰试验
进行胶囊剂溶出度检查时,应取6粒胶囊,尽可 能完全地除尽内容物(起草标准时最好使用未 使用过的同批号的胶囊壳),置同一容器中用 该品种项下规定体积的溶出介质溶解空胶囊壳, 并按照规定的分析方法测定,做必要的校正, 如果校正值不大于标示量的2%,可忽略不计, 如校正值低于标示量的25%,可进行校正,如 校正值大于标示量的25%,试验无效。
药品检验标准操作规程(二)
北京市药品检验所抗生素室 张洁萍
一、制剂
(一)片剂的重量差异 1.简述 在片剂生产中,由于颗粒的均匀度和流动 性,以及工艺、设备和管理等原因,都会 引起片剂重量差异,本项检查的目的在于 控制各片重量的一致性,保证用药剂量的 准确。 凡规定检查含量均匀度的片剂,一般不再 进行该项检查。
4.取连续测定三次结果的平均值。
pH值的测定
1.测定之前,按各品种项下的规定,选择两种标 准缓冲液(pH值相差约3个单位),使供试品 溶液的pH值处于二者之间。
注射用无菌粉末

粉针系用无菌操作法将经过无菌精制的药物分(灌)装于无菌容器中,临用前再用灭菌的注射用溶媒溶解或混悬而制成的剂型。
凡遇热或遇水不稳定的药物如青霉素G、辅酶A、胰蛋白酶等均需制成粉针。
根据制备方法不同,粉针分为注射用无菌粉末和注射用冻干制品两类。
注射用无菌粉末(一)概述注射用无菌粉末又称粉针,临用前用灭菌注射用水溶解后注射,是一种较常用的注射剂型。
适用于在水中不稳定的药物,特别是对湿热敏感的抗生素及生物制品。
依据生产工艺不同,可分为注射用冷冻干燥制品和注射用无菌分装产品。
前者是将灌装了药液的安瓿进行冷冻干燥后封口而得,常见于生物制品,如辅酶类;后者是将已经用灭菌溶剂法或喷雾干燥法精制而得的无菌药物粉末在避菌条件下分装而得,常见于抗生素药品,如青霉素。
由于多数情况下,制成粉针的药物稳定性较差,因此,粉针的制造一般没有灭菌的过程,因而对无菌操作有较严格的要求,特别在灌封等关键工序,最好采用层流洁净措施,以保证操作环境的洁净度。
(二)注射用冻干制品1.冻干无菌粉末的制备工艺由冷冻干燥原理可知,冻干粉末的制备工艺可以分为预冻、减压、升华、干燥等几个过程。
此外,药液在冻干前需经过滤、灌装等处理过程。
(1)预冻:是恒压降温过程。
药液随温度的下降冻结成固体,温度一般应降至产品共熔点以下10~20℃以保证冷冻完全。
若预冻不完全,在减压过程中可能产生沸腾冲瓶的现象,使制品表面不平整。
(2)升华干燥:首先是恒温减压过程,然后是在抽气条件下,恒压升温,使固态水升华逸去。
升华干燥法分为两种,一种是一次升华法,适用于共熔点为-l0~-20℃的制品,且溶液黏度不大。
它首先将预冻后的制品减压,待真空度达一定数值后,启动加热系统缓缓加热,使制品中的冰升华,升华温度约为-20℃,药液中的水分可基本除尽。
另一种是反复冷冻升华法,该法的减压和加热升华过程与一次升华法相同,只是预冻过程须在共熔点与共熔点以下20℃之间反复升降预冻。
而不是一次降温完成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
注射用无菌粉末装量差异检查法标准操作规程
目的:
建立注射用无菌粉末装量差异检查法标准操作规程,控制各瓶间装量的一致性,以保证使用剂量的准确。
2.依据:
《中华人民共和国药典》2000年版二部。
3. 范围:
适用于橡皮塞铝盖玻瓶或安瓿装的注射用无菌粉末的装量差异检查。
4. 职责:
QA检查员、QC检验员对本标准的实施负责。
5. 程序:
5.1. 仪器与用具:分析天平感量1mg或0.1mg(适用于检查装量小于0.1g的粉针剂)
5.2.检查法:
5.2.1. 取供试品5瓶(支),除去铝盖和瓶签(若为纸标签,用水润湿后除去纸屑;
若为直接在玻璃上印字标签,用适当有机溶媒擦除字迹),容器外壁用乙醇洗净,置干燥器内放置1-2小时,俟干燥后,分别编号,依次放于固定位置。
5.2.2.轻扣橡皮塞或安瓿颈,使其上附着的粉末全部落下,分别精密称定每瓶(支)的重量,开启容器(注意避免玻璃屑等异物落入容器中),倾出内容物,容器用水、乙
醇洗净,依次放回原固定位置,在适当的条件下干燥后,再分别精密称定每一容器的重量,即可求出每1瓶(支)的装量和平均装量。
5.2.3.复试、初试中,如有1瓶的装量超过装量差异限度规定时,另取10瓶(支)
按5.2.1.、5.2.2.项下复试。
5.3.记录与计算:
5.3.1. 记录每次称量数据。
5.3.2.根据每瓶(支)的重量与其空瓶重之差,求算每瓶(支)内容物重量。
5.3.3.每瓶(支)内容物重量之和除以5(复试时除以10),即得平均装量( m ),
保留3位有效数字。
5.3.4.按下表规定装量差异限度,求出允许装量范围(m±m×装量差异限度)。
注射用无菌粉末装量差异限度规定
5.4.结果与判定:
5.4.1. 每1瓶(支)中的装量与平均装量相比较,均末超过者,判为符合规定。
5.4.2.每1瓶(支)中的装量与平均装量相比较,超过1瓶者,判为不符合规定。
5.4.3.初试结果仅有1瓶(支)的装量超过允许装量范围时,另取10瓶(支)复
试。
复试结果每1瓶(支)的装量与其允许装量范围相比较,均末超过者,可判为符合规定;若有1瓶(支)或1瓶(支)以上超过时,判为不符合规定。
5.5.注意事项:
5.5.1. 开启安瓿装粉针时,应避免玻璃屑落入或溅失。
5.5.2.用水、乙醇洗涤倾去内容物后的容器时,慎勿将瓶外编号的字迹擦掉,以免影响称量结果;并将空容器与原橡皮塞或安瓿颈部配对放于原固定位置。
5.5.3.空容器的干燥,一般可用60—70℃加热1—2小时,也可在干燥器内干燥
较长时间。
5.5.4.称量空容器时,应注意瓶身与瓶塞(或折断的瓶颈部分)的对号。