欧盟GMP Part I - Basic Requirements for Medicinal Products 第一卷第七章vol4-chap7_2012-06_en
欧盟药品GMP指南

国外药品检查资料汇编GUOW AI Y AOPIN JIANCHA ZILAO HUIBIAN欧盟药品GMP指南OUMENG YAOPINGMP ZHINAN国家食品药品监督管理局药品认证管理中心中国医药科技出版社目录第一部分欧盟药品管理概述 (1)第二部分欧盟GMP基本要求 (33)引言 (35)基本要求I:人用药品及兽药制剂生产质量管理规范 (37)基本要求Ⅱ:原料药生产质量管理规范 (64)第三部分欧盟GMP附录 (103)欧盟GMP附录l无菌药品的生产 (105)欧盟GMP附录2人用生物制品的生产 (119)欧盟GMP附录3放射性药品生产 (126)欧盟GMP附录4兽用非免疫药品的生产 (130)欧盟GMP附录5免疫类兽药制品的生产 (134)欧盟GMP附录6医用气体生产 (143)欧盟GMP附录7草药制剂的生产 (152)欧盟GMP附录8原辅包装材料的取样 (156)欧盟GMP附录9液剂、霜剂和油膏的生产 (160)欧盟GMP附录10定量吸人式气雾剂的生产 (163)欧盟GMP附录ll计算机系统 (166)欧盟GMP附录12药品生产中电离辐射的应用 (170)欧盟GMP附录13临床试验用药的生产 (177)欧盟GMP附录14人血液或血浆制品的生产 (191)欧盟GMP附录15确认和验证...............................................................(19r7) 欧盟GMP附录16药品放行责任人签发证书和放行批产品 (206)欧盟GMP附录17参数放行 (215)欧盟GMP附录19对照样品和留样 (219)欧盟GMP附录20质量风险管理 (224)欧盟GMP术语 (241)第一部分欧盟药品管理概述目录一、欧盟概况 (4)1.欧盟简史 (4)2.体制 (4)3.机构名称 (5)二、欧盟制药业及其产业政策 (6)1.欧洲企业理事总会目标及产业政策 (6)2.制药行业分管机构的目标和使命 (8)三、药品的审评及检查 (9)1.欧洲药品管理局的机构及廉政措施 (9)2.欧洲药品管理局的职能 (11)3.欧盟药事法规 (12)4.产品放行责任人 (15)5.药品审评程序 (17)(1)集中审评程序 (17)(2)互认程序 (20)(3)分散审评程序 (22)6.GXP检查 (22)7.GXP检查问答 (26)四、《欧洲药典》及欧洲药品质量理事会 (27)1.《欧洲药典》 (27)2.欧洲药品质量理事会 (28)3.《欧洲药典》适用性认证 (29)五、药品信息一药品说明书、标签和广告 (29)六、药品安全的持续监控 (30)附录 (32)一、欧盟概况1.欧盟简史二次大战以后,欧洲国家实现团结一致的强烈愿望,以及使欧洲子孙后代有一个和平和稳定的经济发展环境的共同目标,最终导致了欧洲经济共同体机构的产生和演变。
欧盟原料药GMP认证检查项目

欧盟原料药GMP认证检查项目GMP Checklist for Active Pharmaceutical Ingredients(APIs)本GMP 认证检查项目基于法规要求和个人经验。
This GMP checklist is based on regulatory requirements and personal experiences.I. 组织和人员Organization and PersonnelA.质量部门责任Responsibilities of the Quality Unit (QU)C人员资质Personnel QualificationsD人员卫生Personnel HygieneE.顾问ConsultantsII. 厂房和设施Buildings and Facilities A. 设计和结构Design and ConstructionB. 照明C. 通风,空气过滤, 空气加热和控制Ventilation, Air Filtration, Air Heating andControlE. 水暖、洗衣和厕所设施Plumbing, Washing, and Toilet FacilitiesF. 污水和垃圾Sewage and RefuseG. 卫生SanitationH. 维护MaintenanceIII. 工艺设备Process EquipmentA. 设备设计和制造Equipment Design and ConstructionB. 设备校验和确认Equipment Calibration and Qualification汉堡药监局要求有记录册.C. 设备清洁和维护规程Equipment Cleaning and Maintenance ProceduresD. 设备清洁方法Equipment Cleaning MethodsE. 现场清洁方法Clean in Place MethodsF. 自动、机械、电子和计算机设备Automatic, Mechanical, Electronic, andComputer EquipmentIV. 物料管理Materials Management A. 控制通则General ControlsB. 物料的接收、取样、测试和批准Receipt, Sampling, Testing, and Approvalof MaterialsC. 原料的使用和重新评估Use and Re-evaluation of Approved Raw MaterialsD. 原料的拒收Rejected Raw MaterialsE. 回收溶剂,母液,副产物的控制Control of Recovered Solvents, MotherLiquors, and Second CropsF. 工艺用水质量V. 生产和工艺控制Production and Process Controls A. 书面程序和偏差Written Procedures and DeviationsB. 原料的称量和测量Raw Material Weighing and MeasuringC. 收率计算Calculation of YieldD. 设备识别Equipment 标识E. 在线取样和控制In-Process Sampling and ControlsF. 原料药生产的时间限制Time Limits on Production of APIsG. 污染控制Contamination ControlH. 原料药和中间体的混合Blending of APIs and IntermediatesVI. 包装盒标签控制Packaging and Labeling Controls A. 控制通则General ControlsB. 标签的制定和控制Label Issuance and ControlsC. 包装盒贴标操作Packaging and Labeling OperationsD. 包材Packaging MaterialsE. 有效期或复验期Expiration or Retest DatingVII. 储存和分发Storage and Distribution A. 入库程序Warehousing ProceduresB. 分发程序Distribution ProceduresVIII. 实验室控制Laboratory Controls A. 控制通则General ControlsB. 中间体和原料药的检测Testing of Intermediates and APIsC. 稳定性试验Stability TestingD. 留样Reserve / Retention SamplesE. 试验动物Laboratory AnimalsIX. 文件:记录和报告D ocumentation: Records and ReportsA. 控制通则:文件系统和质量标准General Controls: Documentation System and Specifications汉堡药监局要求有记录册B. 设备清洁和使用记录Equipment Cleaning and Use Record汉堡药监局要求有记录册C. 原料、原料药包装和标签材料的记录Records of Raw Materials, API Packaging and Labeling MaterialsD. 主生产和控制记录(生产工艺规程)Master Production and Control Records(Master Production Instructions)E. 批生产和控制记录Batch Production and Control RecordsF. 批生产记录复核Batch Production Record ReviewG. 实验室记录Laboratory Recordsbooks for Sample receipt, Reagents and for equipments calibration and Maintenance.汉堡药监当局要求提供实验室记录册,以及样品接受记录设备校验和维护用的试剂H. 销售记录Distribution RecordsX. 验证ValidationA. 验证方针Validation PolicyB. 验证文件Validation DocumentationC. 确认QualificationD. 工艺验证的方法Approaches to Process ValidationE. 工艺验证程序Process Validation ProgramF. 验证系统的定期审核Periodic Review of Validated SystemsG. 验证清洁方法Validation of Cleaning MethodsH. 分析方法的验证Validation of Analytical MethodsXI. 变更控制Change ControlXII. 拒收和物料的再利用Rejection and Re-Use of Materials A. 拒收RejectionB. 返工ReprocessingC. 重新加工ReworkingD. 物料和溶剂的回收Recovery of Materials and SolventsE. 原料药和中间体的退货Returned APIs and IntermediatesF. 原料药和中间体的回收API and Intermediate SalvagingXIII. 投诉和召回Complaints and RecallsA. 投诉处理Complaint HandlingB. 召回程序Recall Procedures。
(完整版)欧盟GMP附录

欧洲共同体:European Communities (EC)。
欧洲联盟:European Union (EU),简称欧盟。
人用药品注册技术标准国际协调会:ICH欧盟GMP附录1无菌药品的生产注:冻干瓶轧盖的条款自2010年3月1日开始实施。
原则为降低微生物、微粒和热原污染的风险,无菌药品的生产应有各种特殊要求。
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度。
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法和规程进行。
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验。
注:本指南没有对微粒、浮游菌和表面微生物等测试方法详细进行阐述,可参阅欧洲标准或国际标准(CEN/ISO)及药典资料。
总则1.无菌药品的生产必须在洁净区内进行,人员和(或)设备以及物料必须通过缓冲进入洁净区。
洁净区应当保持适当的洁净度,洁净区的送风须经具有一定过滤效率过滤器的过滤。
2.原料配制、产品加工和灌装等不同操作必须在洁净去内彼此分开的单独区域内进行。
生产工艺可分为两类:一类是最终灭菌工艺;第二类是部分或全部工序为无菌操作的工艺。
3.应按所需环境的特点确定无菌产品的洁净级别。
每一步生产操作都应达到适当的动态洁净度,以尽可能降低产品(或原料)被微粒或微生物污染。
洁净区的设计必须符合相应的“静态”标准,以达到“动态”的洁净要求。
“静态”是指安装已经完成并已运行,但没有操作人员在场的状态。
“动态”是指生产设施按预定的工艺模式运行并有规定数量的操作人员进行现场操作的状态。
应确定每一洁净室或每组洁净间的“动态”及“静态”标准。
无菌药品生产所需的洁净区一般可分为4个级别:A级:高风险操作区,如:灌装区,放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配/连接操作的区域。
通常用单向流操作台/罩来维护该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
EU-GMP欧盟GMP中文版

优质参考文档欧盟药品管理规则第4卷药品生产质量管理规范佃98版欧洲共同体前言欧洲共同体制药工业在药品的开发,生产和控制过程中保持高标准的质量保证。
上市许可系统保证由有能力的权威机构对药品的安全,质量和有效性是否达到相应的规定进行评估。
生产许可系统保证在欧洲市场上获准销售的药品是由授权的生产商生产,其日常活动由权威机构定期检查。
无论是在欧共体之内销售,还是在欧共体之外销售,所有欧共体的药品生产企业都必须通过生产许可。
有两个药品生产和质量管理指导原则,药品生产和质量管理规范(GMP)和指南来源于两个指导原则,一个是人用药物指导原则(指导原则91/356/EEC)一个是兽用药物指导原则(指导原则91/412/EEC),这两个指导原则1991年被欧共体采纳。
根据这些原则,制定了详细的药品生产和质量管理规范,用于对申请生产许可的企业进行评估和对药品生产企业进行检查的基础。
GMP的原则和详细的指南适用于需要按照第16条75/319/EEC和修改的第24条81/851/EEC要求认证的所有的操作。
也与所有其它大规模药品生产过程,诸如医院负责的临床试验用药的制备有关。
所有的成员国和工业企业本身都同意GMP适用于人用药物的生产,也适用于兽用药物的生产。
在两个附录中对兽用药品和兽用免疫药品的GMP指南做了详细的调整。
指南用章来表述,每章用标题来概括章节的原则内容。
第一章质量管理列出了药品生产的质量保证的基本概念。
后续各章的原则列出了质量保证的目标和提供了足够的让生产商在执行这一原则时所必须考虑的基本要素。
这一指南除了在9个章节中表述了 GMP的基本要素外,还包括一系列附录提供了与之有关的活动的特定范围的细节。
有时几个附录同时使用,如关于无菌制剂,辐射性药物,生化药物的附录。
在附录后还列出了这一指南所使用的术语表•指南的第一版在1989年出版,包括一个无菌药品生产的附录。
第二版在1992年1月出版;欧共体指到原则包括给人用药品和兽用药品的GMP提供原则和指南的欧共体于1991年6月13日颁布的91/356指导原则和1991年7月23日颁布的91/412指导原则。
20100731 欧盟API GMP 中英文对照 CX 20110112
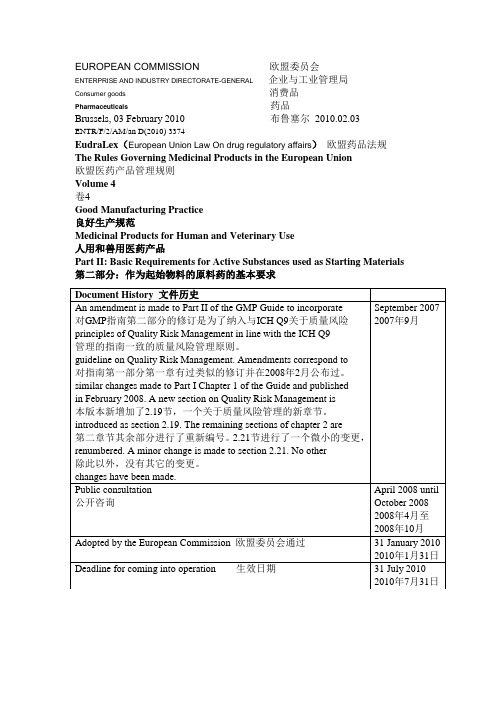
EUROPEAN COMMISSION 欧盟委员会ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL 企业与工业管理局Consumer goods 消费品Pharmaceuticals 药品Brussels, 03 February 2010 布鲁塞尔2010.02.03ENTR/F/2/AM/an D(2010) 3374EudraLex(European Union Law On drug regulatory affairs)欧盟药品法规The Rules Governing Medicinal Products in the European Union欧盟医药产品管理规则Volume 4卷4Good Manufacturing Practice良好生产规范Medicinal Products for Human and Veterinary Use人用和兽用医药产品Part II: Basic Requirements for Active Substances used as Starting Materials 第二部分:作为起始物料的原料药的基本要求Table of Contents目录1 Introduction1简介1.1 Objective1.1目的1.2 Regulatory Applicability1.2法规适用性1.3 Scope1.3范围2 Quality Management2质量管理2.1 Principles2.1原则2.2 Quality Risk Management2.2质量风险管理2.3 Responsibilities of the Quality Unit(s) 2.3质量部门的职责2.4 Responsibility for Production Activities 2.4生产活动的职责2.5 Internal Audits (Self-Inspection)2.5内部审计(自检)2.6 Product Quality Review2.6产品质量回顾3 Personnel3 人员3.1 Personnel Qualifications3.1 人员资质3.2 Personnel Hygiene3.2 人员卫生3.3 Consultants3.3 顾问4 Buildings and Facilities4 厂房设施4.1 Design and Construction4.1 设计和建造4.2 Utilities4.2 公用工程4.3 Water4.3 水4.4 Containment4.4 限制4.5 Lighting4.5 照明4.6 Sewage and Refuse4.6 废水废物4.7 Sanitation and Maintenance4.7 公共卫生及保养5 Process Equipment5 工艺设备5.1 Design and Construction5.1 设计和建造5.2 Equipment Maintenance and Cleaning5.2 设备的保养和清洁5.3 Calibration5.3 校验5.4 Computerized Systems5.4 计算机系统6 Documentation and Records6 文件和记录6.1 Documentation System and Specifications6.1 文件系统与规格标准6.2 Equipment Cleaning and Use Record6.2 设备清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labelling and Packaging Materials 6.3 原料、中间产品、原料药的标签和包装材料的记录6.4 Master Production Instructions (Master Production and Control Records)6.4 生产指令(生产和控制记录)6.5 Batch Production Records (Batch Production and Control Records)6.5批生产记录(批生产和控制记录)6.6 Laboratory Control Records6.6 实验室控制记录(批检验记录)6.7 Batch Production Record Review6.7批生产记录审核7 Materials Management7 物料管理7.1 General Controls7.1 控制通则7.2 Receipt and Quarantine7.2 接受和待检7.3 Sampling and Testing of Incoming Production Materials7.3 到货物料的取样和检测7.4 Storage7.4 贮存7.5 Re-evaluation7.5 再评估8 Production and In-Process Controls8 生产和过程控制8.1 Production Operations8.1 生产操作8.2 Time Limits8.2 时间限制8.3 In-process Sampling and Controls8.3 中控取样和控制8.4 Blending Batches of Intermediates or APIs8.4 中间产品和原料药的混批8.5 Contamination Control8.5 污染控制9 Packaging and Identification Labelling of APIs and Intermediates 9 中间产品和原料药的包装和贴签9.1 General9.1 总则9.2 Packaging Materials9.2 包装材料9.3 Label Issuance and Control9.3 标签放行和控制9.4 Packaging and Labelling Operations9.4 包装和贴签操作10 Storage and Distribution10 贮存和销售10.1 Warehousing Procedures10.1 入库程序10.2 Distribution Procedures10.2 销售程序11 Laboratory Controls11 实验室控制11.1 General Controls11.1 控制通则11.2 Testing of Intermediates and APIs11.2 中间产品和原料药的检测11.3 Validation of Analytical Procedures11.3 分析方法的验证11.4 Certificates of Analysis11.4 分析报告11.5 Stability Monitoring of APIs11.5 原料药的稳定性监测11.6 Expiry and Retest Dating11.6 失效和复检日期11.7 Reserve/Retention Samples11.7 留样12 Validation12 验证12.1 Validation Policy12.1 验证方针12.2 Validation Documentation12.2 验证文件12.3 Qualification12.3 确认12.4 Approaches to Process Validation12.4 工艺验证方法12.5 Process Validation Program12.5 工艺验证计划12.6 Periodic Review of Validated Systems12.6 验证系统的定期审核12.7 Cleaning Validation12.7 清洁验证12.8 Validation of Analytical Methods12.8 分析方法验证13 Change Control13 变更控制14 Rejection and Reuse of Materials14 物料的拒收和再利用14.1 Rejection14.1 拒收14.2 Reprocessing14.2 返工14.3 Reworking14.3 重新加工14.4 Recovery of Materials and Solvents14.4 物料和溶剂的回收利用14.5 Returns14.5 退回15 Complaints and Recalls15 投诉和召回16 Contract Manufacturers (including Laboratories)16 合同生产企业(包含实验室)17 Agents, Brokers, Traders, Distributors, Repackers, and Relabellers 17 代理商、经纪商、贸易商、经销商、重新包装商和重新贴签商17.1 Applicability17.1 适用性17.2 Traceability of Distributed APIs and Intermediates17.2 已销售中间产品和原料药的追踪17.3 Quality Management17.3 质量管理17.4 Repackaging, Relabelling and Holding of APIs and Intermediates 17.4 中间产品和原料药的重新包装、重新贴签和处理17.5 Stability17.5 稳定性17.6 Transfer of Information17.6 信息的传输17.7 Handling of Complaints and Recalls17.7 投诉和召回的处理17.8 Handling of Returns17.8 退货的处理18 Specific Guidance for APIs Manufactured by Cell Culture/Fermentation 18 用于细胞培养/发酵而得原料药的特殊指南18.1 General18.1 总则18.2 Cell Bank Maintenance and Recordkeeping18.2 细胞库的维护和记录保存18.3 Cell Culture/Fermentation18.3 细胞培养/发酵18.4 Harvesting, Isolation, and Purification18.4 收获、分离和精制18.5 Viral Removal/Inactivation Steps18.5 病毒除去/灭火步骤19 APIs for Use in Clinical Trials19 用于临床试验的原料药19.1 General19.1 总则19.2 Quality19.2 质量19.3 Equipment and Facilities19.3 设备设施19.4 Control of Raw Materials19.4 原料的控制19.5 Production19.5 生产19.6 Validation19.6 验证19.7 Changes19.7 变更19.8 Laboratory Controls19.8 实验室控制19.9 Documentation19.9 文件20 Glossary20 词汇表1 Introduction1 介绍This guideline was published in November 2000 as Annex 18 to the GMP Guide reflecting the EU’s agreement to ICH Q7A and has been used by manufacturers and GMP inspectorates on a voluntary basis. Article 46 (f) of Directive 2001/83/EC and Article 50 (f) of Directive 2001/82/EC; as amended by Directives 2004/27/EC and 2004/28/EC respectively, place new obligations on manufacturing authorisation holders to use only active substances that have been manufactured in accordance with Good Manufacturing Practice for starting materials. The directives go on to say that the principles of Good Manufacturing Practice for active substances are to be adopted as detailed guidelines. Member States have agreed that the text of former Annex 18 should form the basis of the detailed guidelines to create Part II of the GMP Guide.本指南已经在2000年11月以GMP指南附录18的形式公布过,它反应了欧盟对ICH Q7A的认可以,该指南已经被生产商和GMP检查员在自愿的原则下所使用。
欧盟GMP(中英文对照)

第一章 质量管理一、原则Principle生产许可证持有厂家只能生产医药产品,以确保药品符合其预期的使用目的,符合销售许可证的要求,并不因药品安全性、质量或药效方面的问题而给患者带来风险。
达到这一质量目标是高层管理者的责任,同时也需要公司各部门、各层次的职员以及公司的供应商和销售商的参与并承担义务。
为了确保达到该质量目标,必须全面设计并正确贯彻实施包括GMP 与质量控制(QC)在内的质量保证(QA)体系。
该体系应用文件明文规定并对其有效性加以监控。
质量保证体系的所有部门都必须充分配备胜任的人员,适宜足够的厂房、设备及设施。
与此同时,生产许可证持有者及受权人员具有另外的法律责任。
The holder of a Manufacturing Authorisation must manufacture medicinal products so as to ensure that they are fit for their intended use, comply with the requirements of the Marketing Authorisation and do not place patients at risk due to inadequate safety, quality or efficacy. The attainment of this quality objective is the responsibility of senior management and requires the participation and commitment by staff in many different departments and at all levels within the company, by the company’s suppliers and by the distributors. To achieve the quality objective in a reliable manner there must be a comprehensively designed and correctly implemented system of Quality Assurance incorporating Good Manufacturing Practice and thus Quality Control. It should be fully documented and its effectiveness monitored. All parts of the Quality Assurance system should be adequately resourced with competent personnel, and suitable and sufficient premises, equipment and facilities. There are additional legal responsibilities for the holder of the Manufacturing Authorisation and for the Qualified Person(s).1.1 质量保证、GMP 和质量控制的基本概念是内在相互联系的。
中国、美国、欧盟GMP中英文版
中华人民共和国卫生部令第 79 号《药品生产质量管理规范(2010年修订)》已于2010年10月19日经卫生部部务会议审议通过,现予以发布,自2011年3月1日起施行。
部长陈竺二○一一年一月十七日第一章总则第一条为规范药品生产质量管理,根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施条例》,制定本规范。
第二条企业应当建立药品质量管理体系。
该体系应当涵盖影响药品质量的所有因素,包括确保药品质量符合预定用途的有组织、有计划的全部活动。
第三条本规范作为质量管理体系的一部分,是药品生产管理和质量控制的基本要求,旨在最大限度地降低药品生产过程中污染、交叉污染以及混淆、差错等风险,确保持续稳定地生产出符合预定用途和注册要求的药品。
第四条企业应当严格执行本规范,坚持诚实守信,禁止任何虚假、欺骗行为。
第二章质量管理第一节原则第五条企业应当建立符合药品质量管理要求的质量目标,将药品注册的有关安全、有效和质量可控的所有要求,系统地贯彻到药品生产、控制及产品放行、贮存、发运的全过程中,确保所生产的药品符合预定用途和注册要求。
第六条企业高层管理人员应当确保实现既定的质量目标,不同层次的人员以及供应商、经销商应当共同参与并承担各自的责任。
第七条企业应当配备足够的、符合要求的人员、厂房、设施和设备,为实现质量目标提供必要的条件。
第二节质量保证第八条质量保证是质量管理体系的一部分。
企业必须建立质量保证系统,同时建立完整的文件体系,以保证系统有效运行。
第九条质量保证系统应当确保:(一)药品的设计与研发体现本规范的要求;(二)生产管理和质量控制活动符合本规范的要求;(三)管理职责明确;(四)采购和使用的原辅料和包装材料正确无误;(五)中间产品得到有效控制;(六)确认、验证的实施;(七)严格按照规程进行生产、检查、检验和复核;(八)每批产品经质量受权人批准后方可放行;(九)在贮存、发运和随后的各种操作过程中有保证药品质量的适当措施;(十)按照自检操作规程,定期检查评估质量保证系统的有效性和适用性。
欧盟GMP现场检查条款
EU cGMP Audit Checklist Page 1EU cGMP AUDIT CHECKLISTDATE: ____________ CLIENT NAME: ____________________________ AUDITORS: __________________________ SITE ADDRESS: __________________________________________________________TYPE of ACTIVITY (cGMP or PAI): ______________________________________________APPLICABLE SECTIONSOBSERVATIONYESNOCOMMENTSIntroductionThe pharmaceutical industry of the European Union maintains high standards of quality assurance in the development, manufacture and control of medicinal products. A system of marketing authorisations ensures that all medicinal products are assessed by a competent authority to ensure compliance with contemporary requirements of safety, quality and efficacy. A system of manufacturing authorisations ensures that all products authorised on the European market are manufactured only by authorised manufacturers, whose activities are regularly inspected by the competent authorities. Manufacturing authorisations are required by all pharmaceuticalmanufacturers in the European Community whether the products are sold within or outside of the Community.Two directives laying down principles and guidelines of good manufacturing practice (GMP) for medicinal products were adopted by the Commission. Directive 2003/94/EC applies to medicinal products for human use and Directive 91/412/EEC for veterinary use. Detailed guidelines in accordance with those principles are published in the Guide to Good Manufacturing Practice, which will be used in assessing applications for manufacturing authorisations and as a basis for inspection of manufacturers of medicinal products.The principles of GMP and the detailed guidelines are applicable to all operations which require the authorisation referred to in Article 40 of Directive 2001/83/EC and in Article 44 ofDirective 2001/82/EC, as amended by Directives 2004/27/EC and 2004/28/EC, respectively. They are also relevant for all other large scale pharmaceutical manufacturing processes, such as that undertaken in hospitals, and for the preparation of products for use in clinical trials. All Member States and the industry agreed that the GMP requirements applicable to the manufacture of veterinary medicinal products are the same as those applicable to themanufacture of medicinal products for human use. Certain detailed adjustments to the GMP guidelines are set out in two annexes specific to veterinary medicinal products and to immunological veterinary medicinal products.The Guide is presented in two parts of basic requirements and specific annexes. Part I covers GMP principles for the manufacture of medicinal products. Part II covers GMP for active substances used as starting materials.EU cGMP Audit ChecklistPage 2Chapters of Part I on “basic requirements” are headed by principles as defined in Directives2003/94/EC and 91/412/EEC. Chapter 1 on Quality Management outlines the fundamental concept of quality assurance as applied to the manufacture of medicinal products. Thereafter, eachchapter has a principle outlining the quality assurance objectives of that chapter and a text which provides sufficient detail for manufacturers to be made aware of the essential matters to be considered when implementing the principle.Part II was newly established on the basis of a guideline developed on the level of ICH and publishe d as ICH Q7a on “active pharmaceutical ingredients”, which was implemented as GMP Annex 18 for voluntary application in 2001. According to the revised Article 47 and Article 51, respectively, of the Directive 2001/83/EC and Directive 2001/82/EC, as amended, detailed guidelines on the principles of GMP for active substances used as starting materials shall be adopted and published by the Commission. The former Annex 18 has been replaced by the new Part II of the GMP Guide, which has an extended application both for the human and the veterinary sector.In addition to the general matters of Good Manufacturing Practice outlined in Parts I and II, a series of annexes providing detail about specific areas of activity is included. For some manufacturing processes, different annexes will apply simultaneously (e.g. annex on sterile preparations and on radiopharmaceuticals and/or on biological medicinal products).A glossary of some terms used in the Guide has been incorporated after the annexes.The Guide is not intended to cover security aspects for the personnel engaged in manufacture. This may be particularly important in the manufacture of certain medicinal products such ashighly active, biological and radioactive medicinal products. However, those aspects are governed by other provisions of Community or national law.Throughout the Guide it is assumed that the requirements of the Marketing Authorisationrelating to the safety, quality and efficacy of the products are systematically incorporated into all the manufacturing, control and release for sale arrangements of the holder of the Manufacturing Authorisation.The manufacture of medicinal products has for many years taken place in accordance with guidelines for Good Manufacturing Practice and the manufacture of medicinal products is not governed by CEN/ISO standards. Harmonised standards as adopted by the European standardisation organisations CEN/ISO may be used at industry’s discretion as a tool forimplementing a quality system in the pharmaceutical sector. The CEN/ISO standards have been considered but the terminology of these standards has not been implemented in this third edition of the Guide.EU cGMP Audit Checklist Page 3It is recognised that there are acceptable methods, other than those described in the Guide, which are capable of achieving the principles of Quality Assurance. The Guide is not intended to place any restraint upon the development of any new concepts or new technologies which have been validated and which provide a level of Quality Assurance at least equivalent to those set out in this Guide. It will be regularly revised.EU cGMP Audit ChecklistPage 4APPLICABLESECTIONSOBSERVATION YES NO COMMENTS 1.0 QualityManagementPrinciples ∙Does the sponsor’s quality management appear to satisfy the criteria outlined in the principlesas detailed in the following statement?The holder of a Manufacturing Authorisation must manufacture medicinal products so as to ensure that theyare fit for their intended use, comply with the requirements of the Marketing Authorisation and do not placepatients at risk due to inadequate safety, quality or efficacy. The attainment of this quality objective is theresponsibility of senior management and requires the participation and commitment by staff in many differentdepartments and at all levels within th e company, by the company’s suppliers and by the distributors. To achievethe quality objective in a reliable manner there must be a comprehensively designed and correctly implementedsystem of Quality Assurance incorporating Good Manufacturing Practice and thus Quality Control. It should befully documented and its effectiveness monitored. All parts of the Quality Assurance system should beadequately resourced with competent personnel, and suitable and sufficient premises, equipment and facilities.There are additional legal responsibilities for the holder of the Manufacturing Authorisation and for theQualified Person(s).Quality Assurance ∙Is the QA group of the firm adhering to the major milestones outlined below?Quality Assurance is a wide ranging concept which covers all matters which individually or collectively influencethe quality of a product. It is the total sum of the organised arrangements made with the object of ensuringthat medicinal products are of the quality required for their intended use. Quality Assurance thereforeincorporates Good Manufacturing Practice plus other factors outside the scope of this Guide. The system ofQuality Assurance appropriate for the manufacture of medicinal products should ensure that:i.medicinal products are designed and developed in a way that takes account of the requirements of GoodManufacturing Practice and Good Laboratory Practice;ii.production and control operations are clearly specified and Good Manufacturing Practice adopted;iii.managerial responsibilities are clearly specified;iv.arrangements are made for the manufacture, supply and use of the correct starting and packaging materials;v.all necessary controls on intermediate products, and any other in-process controls and validations are carried out;vi.the finished product is correctly processed and checked, according to the defined procedures;vii.medicinal products are not sold or supplied before a Qualified Person has certified that each production batch has been produced and controlled in accordance with the requirements of the Marketing Authorisation and any otherregulations relevant to the production, control and release of medicinal products;viii.satisfactory arrangements exist to ensure, as far as possible, that the medicinal products are stored, distributed and subsequently handled so that quality is maintained throughout their shelf life;ix.there is a procedure for Self-Inspection and/or quality audit which regularly appraises the effectiveness andapplicability of the Quality Assurance system.cGMP ∙Does the QA group adhere to cGMP concepts as outlined below?Good Manufacturing Practice is that part of Quality Assurance which ensures that products are consistentlyproduced and controlled to the quality standards appropriate to their intended use and as required by theMarketing Authorisation or product specification. Good Manufacturing Practice is concerned with bothproduction and quality control. The basic requirements of GMP are that:EU cGMP Audit Checklist Page 51.0 Quality Managementi.all manufacturing processes are clearly defined, systematically reviewed in the light of experience and shown to be capable of consistently manufacturing medicinal products of the required quality and complying with their specifications;ii. critical steps of manufacturing processes and significant changes to the process are validated; iii. all necessary facilities for GMP are provided including: iv. appropriately qualified and trained personnel; v. adequate premises and space; vi. suitable equipment and services;vii. correct materials, containers and labels; viii. approved procedures and instructions; ix. suitable storage and transport;x. instructions and procedures are written in an instructional form in clear and unambiguous language, specifically applicable to the facilities provided;xi. operators are trained to carry out procedures correctly;xii.records are made, manually and/or by recording instruments, during manufacture which demonstrate that all thesteps required by the defined procedures and instructions were in fact taken and that the quantity and quality of the product was as expected. Any significant deviations are fully recorded and investigated;xiii. records of manufacture including distribution which enable the complete history of a batch to be traced, are retained in a comprehensible and accessible form;xiv. the distribution (wholesaling) of the products minimises any risk to their quality; xv. a system is available to recall any batch of product, from sale or supply;xvi.complaints about marketed products are examined, the causes of quality defects investigated and appropriate measures taken in respect of the defective products and to prevent reoccurrence.Quality Control∙ Does the QA group adhere to the QC principles outlined below?Quality Control is that part of Good Manufacturing Practice which is concerned with sampling, specifications and testing, and with the organisation, documentation and release procedures which ensure that the necessary and relevant tests are actually carried out and that materials are not released for use, nor products released for sale or supply, until their quality has been judged to be satisfactory. The basic requirements of Quality Control are that:i.adequate facilities, trained personnel and approved procedures are available for sampling, inspecting and testing starting materials, packaging materials, intermediate, bulk, and finished products, and where appropriate for monitoring environmental conditions for GMP purposes;ii. samples of starting materials, packaging materials, intermediate products, bulk products and finished products are taken by personnel and by methods approved by Quality Control; iii. test methods are validated;iv. records are made, manually and/or by recording instruments, which demonstrate that all the required sampling, inspecting and testing procedures were actually carried out. Any deviations are fully recorded and investigated; v.the finished products contain active ingredients complying with the qualitative and quantitative composition of the Marketing Authorisation, are of the purity required, and are enclosed within their proper containers and correctly labelled;vi.records are made of the results of inspection and that testing of materials, intermediate, bulk, and finished products is formally assessed against specification. Product assessment includes a review and evaluation of relevant production documentation and an assessment of deviations from specified procedures;vii. no batch of product is released for sale or supply prior to certification by a Qualified Person that it is in accordance with the requirements of the Marketing Authorisation;viii.sufficient reference samples of starting materials and products are retained to permit future examination of theEU cGMP Audit Checklist Page 61.0 Quality Managementproduct if necessary and that the product is retained in its final pack unless exceptionally large packs are produced.Product QualityReviewDoes the QA group adhere to the following principles regarding product quality review?Regular periodic or rolling quality reviews of all licensed medicinal products, including export only products, should be conducted with the objective of verifying the consistency of the existing process, theappropriateness of current specifications for both starting materials and finished product to highlight any trends and to identify product and process improvements. Such reviews should normally be conducted and documented annually, taking into account previous reviews, and should include at least:i. A review of starting materials and packaging materials used for the product, especially those from new sources; ii. A review of critical in-process controls and finished product results;iii. A review of all batches that failed to meet established specification(s) and their investigation;iv. A review of all significant deviations or non-conformances, their related investigations, and the effectiveness of resultant corrective and preventative actions taken;v. A review of all changes carried out to the processes or analytical methods;vi. A review of Marketing Authorisation variations submitted/granted/refused, including those for third country (export only) dossiers;vii. A review of the results of the stability monitoring programme and any adverse trends;viii. A review of all quality-related returns, complaints and recalls and the investigations performed at the time; ix. A review of adequacy of any other previous product process or equipment corrective actions;x. For new marketing authorisations and variations to marketing authorisations, a review of post-marketing commitments;xi. The qualification status of relevant equipment and utilities, e.g. HVAC, water, compressed gases, etc; xii.A review of Technical Agreements to ensure that they are up to date.The manufacturer and marketing authorisation holder, where different, should evaluate the results of thisreview and an assessment should be made whether corrective and preventative action or any revalidation should be undertaken. Reasons for such corrective actions should be documented. Agreed corrective and preventative actions should be completed in a timely and effective manner. There should be management procedures for the ongoing management and review of these actions and the effectiveness of these procedures verified during self inspection. Quality reviews may be grouped by product type, e.g. solid dosage forms, liquid dosage forms, sterile products, etc. where scientifically justified.Where the marketing authorisation holder is not the manufacturer, there should be a technical agreement in place between the various parties that defines their respective responsibilities in producing the quality review. The Qualified Person responsible for final batch certification together with the marketing authorisation holder should ensure that the quality review is performed in a timely manner and is accurate.EU cGMP Audit Checklist Page 7APPLICABLE SECTIONS OBSERVATIONYESNOCOMMENTS2.0 PersonnelPrinciple∙ Does the sponsor have adequate personnel systems in place to meet the criteria outlined in the following statement?The establishment and maintenance of a satisfactory system of quality assurance and the correct manufacture of medicinal products relies upon people. For this reason there must be sufficient qualified personnel to carry out all the tasks which are the responsibility of the manufacturer. Individual responsibilities should be clearly understood by the individuals and recorded. All personnel should be aware of the principles of Good Manufacturing Practice that affect them and receive initial and continuing training, including hygiene instructions, relevant to their needs.General∙ Is the sponsor in compliance with the following statements?2.1 The manufacturer should have an adequate number of personnel with the necessary qualifications and practical experience. The responsibilities placed on any one individual should not be so extensive as to present any risk to quality.2.2 The manufacturer must have an organisation chart. People in responsible positions should have specific duties recorded in written job descriptions and adequate authority to carry out their responsibilities. Their duties may be delegated to designated deputies of a satisfactory qualification level. There should be no gaps or unexplained overlaps in the responsibilities of those personnel concerned with the application of Good Manufacturing Practice.Key Personnel∙ Is the sponsor in compliance with the following areas?2.3 Key Personnel include the head of Production, the head of Quality Control, and if at least one of these persons is not responsible for the duties described in Article 51 of Directive 2001/83/EC,1 the QualifiedPerson(s) designated for the purpose. Normally key posts should be occupied by full-time personnel. The heads of Production and Quality Control must be independent from each other. In large organisations, it may be necessary to delegate some of the functions listed in 2.5, 2.6 and 2.7.2.4 The duties of the Qualified Person(s) are fully described in Article 51 of Directive 2001/83/EC, and can be summarised as follows:a. for medicinal products manufactured within the European Community, a Qualified Person must ensurethat each batch has been produced and tested/checked in accordance with the directives and the marketing authorisation;2 b. for medicinal products manufactured outside the European Community, a Qualified Person mustensure that each imported batch has undergone, in the importing country, the testing specified in paragraph 1 (b) of Article 51; c. a Qualified Person must certify in a register or equivalent document, as operations are carried outand before any release, that each production batch satisfies the provisions of Article 51.The persons responsible for these duties must meet the qualification requirements laid down in Article 493 of2.0 Personnelthe same Directive, they shall be permanently and continuously at the disposal of the holder of theManufacturing Authorisation to carry out their responsibilities. Their responsibilities may be delegated, butonly to other Qualified Person(s).2.5 The head of the Production Department generally has the following responsibilities:i.to ensure that products are produced and stored according to the appropriate documentation inorder to obtain the required quality;ii.to approve the instructions relating to production operations and to ensure their strictimplementation;iii.to ensure that the production records are evaluated and signed by an authorised person before theyare sent to the Quality Control Department;iv.to check the maintenance of his department, premises and equipment;v.to ensure that the appropriate validations are done;vi.to ensure that the required initial and continuing training of his department personnel is carried outand adapted according to need.2.6 The head of the Quality Control Department generally has the following responsibilities:i.to approve or reject, as he sees fit, starting materials, packaging materials, and intermediate, bulkand finished products;ii.to evaluate batch records;iii.to ensure that all necessary testing is carried out;iv.to approve specifications, sampling instructions, test methods and other Quality Control procedures;v.to approve and monitor any contract analysts;vi.to check the maintenance of his department, premises and equipment;vii.to ensure that the appropriate validations are done;viii.to ensure that the required initial and continuing training of his department personnel is carried outand adapted according to need.Other duties of the Quality Control Department are summarised in Chapter 6.2.7 The heads of Production and Quality Control generally have some shared, or jointly exercised,responsibilities relating to quality. These may include, subject to any national regulations:∙the authorisation of written procedures and other documents, including amendments;∙the monitoring and control of the manufacturing environment;∙plant hygiene;∙process validation;∙training;∙the approval and monitoring of suppliers of materials;∙the approval and monitoring of contract manufacturers;∙the designation and monitoring of storage conditions for materials and products;EU cGMP Audit Checklist Page 8EU cGMP Audit Checklist Page 92.0 Personnel∙ the retention of records;∙ the monitoring of compliance with the requirements of Good Manufacturing Practice;∙the inspection, investigation, and taking of samples, in order to monitor factors which may affect product quality.Training∙ Is the sponsor in compliance with the following areas?2.8 The manufacturer should provide training for all the personnel whose duties take them into production areas or into control laboratories (including the technical, maintenance and cleaning personnel), and for other personnel whose activities could affect the quality of the product.2.9 Besides the basic training on the theory and practice of Good Manufacturing Practice, newly recruited personnel should receive training appropriate to the duties assigned to them. Continuing training should also be given, and its practical effectiveness should be periodically assessed. Training programmes should be available, approved by either the head of Production or the head of Quality Control, as appropriate. Training records should be kept.2.10 Personnel working in areas where contamination is a hazard, e.g. clean areas or areas where highly active, toxic, infectious or sensitising materials are handled, should be given specific training.2.11 Visitors or untrained personnel should, preferably, not be taken into the Production and Quality Control areas. If this is unavoidable, they should be given information in advance, particularly about personal hygiene and the prescribed protective clothing. They should be closely supervised.2.12 The concept of Quality Assurance and all the measures capable of improving its understanding and implementation should be fully discussed during the training sessions.Personnel Hygiene∙ Is the sponsor in compliance with the following areas?2.13 Detailed hygiene programmes should be established and adapted to the different needs within the factory. They should include procedures relating to the health, hygiene practices and clothing of personnel.These procedures should be understood and followed in a very strict way by every person whose duties take him into the production and control areas. Hygiene programmes should be promoted by management and widely discussed during training sessions.2.14 All personnel should receive medical examination upon recruitment. It must be the manufacturer’sresponsibility that there are instructions ensuring that health conditions that can be of relevance to the quality of products c ome to the manufacturer’s knowledge. After the first medical examination, examinations should be carried out when necessary for the work and personal health.2.15 Steps should be taken to ensure as far as is practicable that no person affected by an infectious disease or having open lesions on the exposed surface of the body is engaged in the manufacture of medicinal products.2.16 Every person entering the manufacturing areas should wear protective garments appropriate to the operations to be carried out.2.0 Personnel2.17 Eating, drinking, chewing or smoking, or the storage of food, drink, smoking materials or personalmedication in the production and storage areas should be prohibited. In general, any unhygienic practice withinthe manufacturing areas or in any other area where the product might be adversely affected, should beforbidden.2.18 Direct contact should be avoided between the operator’s hands and the exposed product as well as withany part of the equipment that comes into contact with the products.2.19 Personnel should be instructed to use the hand-washing facilities.2.20 Any specific requirements for the manufacture of special groups of products, for example sterilepreparations, are covered in the annexes.EU cGMP Audit Checklist Page 10EU cGMP Audit Checklist Page 11APPLICABLE SECTIONS OBSERVATIONYESNOCOMMENTS3.0 Premises & EquipmentPrinciple∙ Is the sponsor in compliance with the following areas?Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build up of dust or dirt and, in general, any adverse effect on the quality of products.Premises∙ Is the sponsor in compliance with the following areas?General3.1 Premises should be situated in an environment which, when considered together with measures to protect the manufacture, presents minimal risk of causing contamination of materials or products.3.2 Premises should be carefully maintained, ensuring that repair and maintenance operations do not present any hazard to the quality of products. They should be cleaned and, where applicable, disinfected according to detailed written procedures.3.3 Lighting, temperature, humidity and ventilation should be appropriate and such that they do not adversely affect, directly or indirectly, either the medicinal products during their manufacture and storage, or the accurate functioning of equipment.3.4 Premises should be designed and equipped so as to afford maximum protection against the entry of insects or other animals.3.5 Steps should be taken in order to prevent the entry of unauthorised people. Production, storage and quality control areas should not be used as a right of way by personnel who do not work in them.Production Area3.6 In order to minimise the risk of a serious medical hazard due to cross-contamination, dedicated and self contained facilities must be available for the production of particular medicinal products, such as highlysensitising materials (e.g. penicillins) or biological preparations (e.g. from live micro-organisms). The production of certain additional products, such as certain antibiotics, certain hormones, certain cytotoxics, certain highly active drugs and non-medicinal products should not be conducted in the same facilities. For those products, in exceptional cases, the principle of campaign working in the same facilities can be accepted provided thatspecific precautions are taken and the necessary validations are made. The manufacture of technical poisons, such as pesticides and herbicides, should not be allowed in premises used for the manufacture of medicinal products.3.7 Premises should preferably be laid out in such a way as to allow the production to take place in areas connected in a logical order corresponding to the sequence of the operations and to the requisite cleanliness levels.。
(完整版)欧盟GMP附录
欧洲共同体:European Communities (EC)。
欧洲联盟:European Union (EU),简称欧盟。
人用药品注册技术标准国际协调会:ICH欧盟GMP附录1无菌药品的生产注:冻干瓶轧盖的条款自2010年3月1日开始实施。
原则为降低微生物、微粒和热原污染的风险,无菌药品的生产应有各种特殊要求。
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度。
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法和规程进行。
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验。
注:本指南没有对微粒、浮游菌和表面微生物等测试方法详细进行阐述,可参阅欧洲标准或国际标准(CEN/ISO)及药典资料。
总则1.无菌药品的生产必须在洁净区内进行,人员和(或)设备以及物料必须通过缓冲进入洁净区。
洁净区应当保持适当的洁净度,洁净区的送风须经具有一定过滤效率过滤器的过滤。
2.原料配制、产品加工和灌装等不同操作必须在洁净去内彼此分开的单独区域内进行。
生产工艺可分为两类:一类是最终灭菌工艺;第二类是部分或全部工序为无菌操作的工艺。
3.应按所需环境的特点确定无菌产品的洁净级别。
每一步生产操作都应达到适当的动态洁净度,以尽可能降低产品(或原料)被微粒或微生物污染。
洁净区的设计必须符合相应的“静态”标准,以达到“动态”的洁净要求。
“静态”是指安装已经完成并已运行,但没有操作人员在场的状态。
“动态”是指生产设施按预定的工艺模式运行并有规定数量的操作人员进行现场操作的状态。
应确定每一洁净室或每组洁净间的“动态”及“静态”标准。
无菌药品生产所需的洁净区一般可分为4个级别:A级:高风险操作区,如:灌装区,放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配/连接操作的区域。
通常用单向流操作台/罩来维护该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
欧盟GMP(EUGMP)中文版欧洲药品生产和质量管理规范附录1,无菌药品生产
欧盟GMP(EUGMP)中文版欧洲药品生产和质量管理规范附录1,无菌药品生产盟欧盟 GMP cfu/4 小时 cfu/碟5 指手套cfu/手套A <1 <1 <1 <1B 10 5 5 5C 100 50 25 -D 200 100 50 -注:(a)表中各数值均为平均值。
(b)单个沉降碟的暴露时间可以少于 4 小时。
6.应当对微粒和微生物监控制定适当的警戒和纠偏标准。
操作规程中应详细说明结果超标时应采取的纠偏措施。
隔离技术 7.采用能最大限度降地低生产区人员影响的隔离技术,可大大降低无菌生产中环境对产品微生物污染的风险。
隔离操作台和传递装置的设计可以有多种形式。
隔离操作台及其所处环境的设计,应能保证相应区域空气的质量达到设定标准。
隔离操作台所采用的材料在某种程度上易被穿剌或易产生渗漏。
传输装置可设计成单门的、双门的,甚至可以是同灭菌设备相连的全密封系统。
将物品放入隔离操作台或从中取出属污染风险最为严重的操作过程。
尽管人们认为这类隔离操作器的工作区内不一定要有层流,但是,隔离系统通常是用于进行高污染风险操作的场所。
隔离操作台所处环境的级别取决于它们的设计及其应用。
无菌操作的隔离操作台所处环境的级别应予控制,至少为 D 级。
8.隔离操作台只有经过适当的验证之后方可投入使用。
验证时应当考虑到隔离技术的所有关键性因素,例如,隔离系统内部和外部(所处环境)的空气质量、隔离操作台的消毒、传递操作以及隔离系统的完好性。
9.隔离操作器和隔离用袖管/手套系统应进行常规监测,这包括经常进行必要的检漏试验。
吹气/灌装/密封技术 10.吹气/灌装/密封系统是一套专用机械设备,连续操作,从一个热塑性颗粒吹制成容器至灌装和密封,整个过程由一台全自动机器完成。
用于无菌生产的吹气/灌装/密封设备本身装有 A 级空气风淋装置,在操作人员按A/B 级区要求着装的条件下,该设备可以安装在洁净度至少为C 级的环境中。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Ref. Ares(2012)778531 - 28/06/2012 EUROPEAN COMMISSIONHEALTH AND CONSUMERS DIRECTORATE-GENERALHealth Systems and ProductsMedicinal Products - Quality, safety and efficacyBrussels,SANCO/AM/sl/ddg1.d.6(2012)860362EudraLexThe Rules Governing Medicinal Products in the European UnionVolume 4EU Guidelines forGood Manufacturing Practice forMedicinal Products for Human and Veterinary UseChapter 7Outsourced ActivitiesLegal basis for publishing the detailed guidelines: Article 47 of Directive2001/83/EC on the Community code relating to medicinal products for human use andArticle 51 of Directive 2001/82/EC on the Community code relating to veterinary medicinal products. This document provides guidance for the interpretation of the principles and guidelines of good manufacturing practice (GMP) for medicinalproducts as laid down in Directive 2003/94/EC for medicinal products for human useand Directive 91/412/EEC for veterinary use.Status of the document: revision 1Reasons for changes: In view of the ICH Q10 guideline on the PharmaceuticalQuality System, Chapter 7 of the GMP Guide has been revised in order to provideupdated guidance on outsourced GMP regulated activities beyond the current scope ofcontract manufacture and analysis operations. The title of the Chapter has beenchanged to reflect this.Deadline for coming into operation: 31 January 2013Commission Européenne, B-1049 Bruxelles / Europese Commissie, B-1049 Brussel – Belgium. Telephone: (32-2) 299 11 11 PrincipleAny activity covered by the GMP Guide that is outsourced should be appropriately defined, agreed and controlled in order to avoid misunderstandings which could result in a product or operation of unsatisfactory quality. There must be a written Contract between the Contract Giver and the Contract Acceptor which clearly establishes the duties of each party. The Quality Management System of the Contract Giver must clearly state the way that the Qualified Person certifying each batch of product for release exercises his full responsibility.Note: This Chapter deals with the responsibilities of manufacturers towards the Competent Authorities of the Member States with respect to the granting of marketing and manufacturing authorizations. It is not intended in any way to affect the respective liability of Contract Acceptors and Contract Givers to consumers; this is governed by other provisions of Community and national law.General7.1 There should be a written Contract covering the outsourced activities, theproducts or operations to which they are related, and any technical arrangements made in connection with it.7.2 All arrangements for the outsourced activities including any proposed changesin technical or other arrangements should be in accordance with regulations in force, and the Marketing Authorisation for the product concerned, where applicable.7.3 Where the marketing authorization holder and the manufacturer are not thesame, appropriate arrangements should be in place, taking into account the principles described in this chapter.The Contract Giver7.4 The pharmaceutical quality system of the Contract Giver should include thecontrol and review of any outsourced activities. The Contract Giver is ultimately responsible to ensure processes are in place to assure the control of outsourced activities. These processes should incorporate quality risk management principles and notably include:7.5 Prior to outsourcing activities, the Contract Giver is responsible for assessingthe legality, suitability and the competence of the Contract Acceptor to carry out successfully the outsourced activities. The Contract Giver is also responsible for ensuring by means of the Contract that the principles and guidelines of GMP as interpreted in this Guide are followed.7.6 The Contract Giver should provide the Contract Acceptor with all theinformation and knowledge necessary to carry out the contracted operations correctly in accordance with regulations in force, and the Marketing Authorisation for the product concerned. The Contract Giver should ensure that the Contract Acceptor isfully aware of any problems associated with the product or the work which might pose a hazard to his premises, equipment, personnel, other materials or other products.7.7 The Contract Giver should monitor and review the performance of the Contract Acceptor and the identification and implementation of any needed improvement.7.8 The Contract Giver should be responsible for reviewing and assessing the records and the results related to the outsourced activities. He should also ensure, either by himself, or based on the confirmation of the Contract Acceptor’s Qualified Person, that all products and materials delivered to him by the Contract Acceptor have been processed in accordance with GMP and the marketing authorisation.The Contract Acceptor7.9 The Contract Acceptor must be able to carry out satisfactorily the work ordered by the Contract Giver such as having adequate premises, equipment, knowledge, experience, and competent personnel.7.10 The Contract Acceptor should ensure that all products, materials and knowledge delivered to him are suitable for their intended purpose.7.11 The Contract Acceptor should not subcontract to a third party any of the work entrusted to him under the Contract without the Contract Giver’s prior evaluation and approval of the arrangements. Arrangements made between the Contract Acceptor and any third party should ensure that information and knowledge, including those from assessments of the suitability of the third party, are made available in the same way as between the original Contract Giver and Contract Acceptor.7.12 The Contract Acceptor should not make unauthorized changes, outside the terms of the Contract, which may adversely affect the quality of the outsourced activities for the Contract Giver.7.13 The Contract Acceptor should understand that outsourced activities, including contract analysis, may be subject to inspection by the competent authorities.The Contract7.14 A Contract should be drawn up between the Contract Giver and the Contract Acceptor which specifies their respective responsibilities and communication processes relating to the outsourced activities. Technical aspects of the Contract should be drawn up by competent persons suitably knowledgeable in related outsourced activities and Good Manufacturing Practice. All arrangements for outsourced activities must be in accordance with regulations in force and the Marketing Authorisation for the product concerned and agreed by both parties.7.15 The Contract should describe clearly who undertakes each step of the outsourced activity, e.g. knowledge management, technology transfer, supply chain, subcontracting, quality and purchasing of materials, testing and releasing materials,undertaking production and quality controls (including in-process controls, sampling and analysis).7.16 All records related to the outsourced activities, e.g. manufacturing, analytical and distribution records, and reference samples, should be kept by, or be available to, the Contract Giver. Any records relevant to assessing the quality of a product in the event of complaints or a suspected defect or to investigating in the case of a suspected falsified product must be accessible and specified in the relevant procedures of the Contract Giver.7.17 The Contract should permit the Contract Giver to audit outsourced activities, performed by the Contract Acceptor or his mutually agreed subcontractors。