第一类医疗器械医用帽备案资料模板-3、产品 技术要求
第一类医疗器械产品备案资料要求及说明

第一类医疗器械产品备案资料要求及说明第一类医疗器械是指对人体进行诊断、预防、监测、治疗或减轻疾病等方面具有辅助作用的器械。
根据我国《医疗器械监督管理条例》,第一类医疗器械的备案是指将医疗器械产品的基本信息进行登记备案,并按照相关要求制作、提交相应备案材料。
1.备案申请书:申请者应填写《医疗器械备案登记申请书》,包括申请单位基本信息、产品基本信息等。
2.产品的技术要求:产品的技术要求应符合国家法律法规的相关规定,包括产品的功能、性能、使用方法、材料组成等详细说明。
3.产品的标识与说明书:申请者应提供产品的标识图样和规格,并提供产品说明书,说明产品的使用方法、适应范围、使用限制等。
4.产品的样品:申请者应提供产品的样品,以供审查和测试。
5.检验报告:申请者应提供由具备资质的检验机构出具的产品检验报告,报告中应包括产品的性能测试结果、安全性评价等相关内容。
6.生产质量管理规范:申请者应提供生产质量管理规范文件,包括产品原材料供应管理、产品生产工艺流程管理、产品质量控制体系等。
7.其他相关材料:视具体情况可以要求申请者提供产品的临床试验报告、国际标准与技术规范、产品的专利证书等。
以上是第一类医疗器械备案所需的主要资料要求。
备案申请者需要确保提供的资料真实、准确,并保证符合国家法律法规和相关技术要求。
此外,备案申请者还需注意以下几点说明:1.备案资料的提供:备案申请人要完整、准确地提供所有备案所需的相关资料,确保内容完整、规范、合理。
2.技术要求的符合:备案申请人要仔细阅读国家法律法规和相关技术要求,确保产品的技术要求符合规定,否则可能会影响备案的通过。
3.检验报告的准确性:备案申请者应选择具备资质的检验机构进行产品的检验测试,并确保检验报告内容真实、准确。
4.备案申请时间:备案申请者应提前了解备案所需的时间周期,并提前进行备案申请,以确保在产品上市前完成备案。
总之,第一类医疗器械备案是一项非常重要的程序,备案资料的准备和审查过程应严格按照国家法律法规和相关要求进行,在备案过程中要审慎操作、保护好备案资料的机密性,以确保备案的顺利进行。
NMPA第一类医疗器械备案资料要求及说明

附1第一类医疗器械备案资料要求及说明一、备案资料(一)第一类医疗器械备案表(二)关联文件1.境内备案人提供:企业营业执照副本或事业单位法人证书的复印件。
委托其他企业生产的,应当提供受托企业资格文件(营业执照副本复印件)、委托合同和质量协议。
2.境外备案人提供:(1)境外备案人企业资格证明文件:境外备案人注册地所在国家(地区)公司登记主管部门或医疗器械主管部门出具的能够证明境外备案人存续且具备相应医疗器械生产资格的证明文件;或第三方认证机构为境外备案人出具的能够证明境外备案人具备相应医疗器械生产资格的证明文件。
(2)境外备案人注册地或生产地址所在国家(地区)医疗器械主管部门出具的准许该产品上市销售的证明文件。
备案人注册地或生产地址所在国家(地区)未将该产品作为医疗器械管理的,备案人需提供相关文件,包括备案人注册地或者生产地所在国家(地区)准许该产品上市销售的证明文件。
未在境外备案人注册地或生产地所在国家(地区)上市的创新医疗器械可以不提交。
(3)境外备案人在中国境内指定代理人的委托书、代理人承诺书,代理人营业执照副本复印件。
(三)产品技术要求产品技术要求应按照《医疗器械产品技术要求编写指导原则》编制,主要包括医疗器械成品的可进行客观判定的功能性、安全性指标和检测方法。
(四)产品检验报告产品检验报告应为产品全性能自检报告或委托检验报告,检验的产品应当具有典型性。
(五)产品说明书及最小销售单元标签设计样稿说明书和标签应当符合《医疗器械说明书和标签管理规定》的相关要求,说明书中产品性能应当与产品技术要求中的相应内容一致。
进口产品应当提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
(六)生产制造信息对生产过程相关情况的概述。
无源医疗器械应明确产品生产加工工艺,注明关键工艺和特殊工艺。
有源医疗器械应提供产品生产工艺过程的描述性资料,可采用流程图的形式,是生产过程的概述。
体外诊断试剂应概述主要生产工艺,包括:固相载体、显色系统等的描述及确定依据,反应体系包括样本采集及处理、样本要求、样本用量、试剂用量、反应条件、校准方法(如果需要)、质控方法等。
一类医疗器械备案产品技术要求编写内容和格式模板

产品技术要求编写内容和格式产品技术要求应按照《医疗器械产品技术要求编写指导原则》编制。
医疗器械产品技术要求编写指导原则根据《医疗器械监督管理条例》等相关规定,制定本指导原则。
一、基本要求(一)医疗器械产品技术要求的编制应符合国家相关法律法规。
(二)医疗器械产品技术要求中应采用规范、通用的术语。
如涉及特殊的术语,需提供明确定义,并写到“4.术语”部分。
(三)医疗器械产品技术要求中的检验方法各项内容的编号原则上应和性能指标各项内容的编号相对应。
(四)医疗器械产品技术要求中的文字、数字、公式、单位、符号、图表等应符合标准化要求。
(五)如医疗器械产品技术要求中的内容引用国家标准、行业标准或中国药典,应保证其有效性,并注明相应标准的编号和年号以及中国药典的版本号。
二、内容要求医疗器械产品技术要求的内容应符合以下要求:(一)产品名称。
产品技术要求中的产品名称应使用中文,并与申请注册(备案)的中文产品名称相一致。
(二)产品型号/规格及其划分说明。
产品技术要求中应明确产品型号和/或规格,以及其划分的说明。
对同一注册单元中存在多种型号和/或规格的产品,应明确各型号及各规格之间的所有区别(必要时可附相应图示进行说明)。
对于型号/规格的表述文本较大的可以附录形式提供。
(三)性能指标。
1. 产品技术要求中的性能指标是指可进行客观判定的成品的功能性、安全性指标以及质量控制相关的其他指标。
产品设计开发中的评价性内容(例如生物相容性评价)原则上不在产品技术要求中制定。
2. 产品技术要求中性能指标的制定应参考相关国家标准/行业标准并结合具体产品的设计特性、预期用途和质量控制水平且不应低于产品适用的强制性国家标准/行业标准。
3. 产品技术要求中的性能指标应明确具体要求,不应以“见随附资料”、“按供货合同”等形式提供。
(四)检验方法。
检验方法的制定应与相应的性能指标相适应。
应优先考虑采用公认的或已颁布的标准检验方法。
检验方法的制定需保证具有可重现性和可操作性,需要时明确样品的制备方法,必要时可附相应图示进行说明,文本较大的可以附录形式提供。
第一类医疗器械备案资料要求及说明

附件1第一类医疗器械备案资料要求及说明一、备案资料(一)第一类医疗器械备案表(二)安全风险分析报告医疗器械应按照YY 0316《医疗器械风险管理对医疗器械的应用》的有关要求编制,主要包括医疗器械预期用途和与安全性有关特征的判定、危害的判定、估计每个危害处境的风险;对每个已判定的危害处境,评价和决定是否需要降低风险;风险控制措施的实施和验证结果,必要时应引用检测和评价性报告;任何一个或多个剩余风险的可接受性评定等,形成风险管理报告。
体外诊断试剂应对产品寿命周期的各个环节,从预期用途、可能的使用错误、与安全性有关的特征、已知和可预见的危害等方面的判定及对患者风险的估计进行风险分析、风险评价及相应的风险控制的基础上,形成风险管理报告。
(三)产品技术要求产品技术要求应按照《医疗器械产品技术要求编写指导原则》编制。
(四)产品检验报告产品检验报告应为产品全性能自检报告或委托检验报告,检验的产品应当具有典型性。
(五)临床评价资料1. 详述产品预期用途,包括产品所提供的功能,并可描述其适用的医疗阶段(如治疗后的监测、康复等),目标用户及其操作该产品应具备的技能/知识/培训;预期与其组合使用的器械。
2. 详述产品预期使用环境,包括该产品预期使用的地点如医院、医疗/临床实验室、救护车、家庭等,以及可能会影响其安全性和有效性的环境条件(如温度、湿度、功率、压力、移动等)。
3. 详述产品适用人群,包括目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息,以及使用过程中需要监测的参数、考虑的因素。
4. 详述产品禁忌症,如适用,应明确说明该器械禁止使用的疾病或情况。
5. 已上市同类产品临床使用情况的比对说明。
6. 同类产品不良事件情况说明。
(六)产品说明书及最小销售单元标签设计样稿医疗器械应符合相应法规规定。
进口医疗器械产品应提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
体外诊断试剂产品应按照《体外诊断试剂说明书编写指导原则》的有关要求,并参考有关技术指导原则编写产品说明书。
《第一类医疗器械备案》材料要求
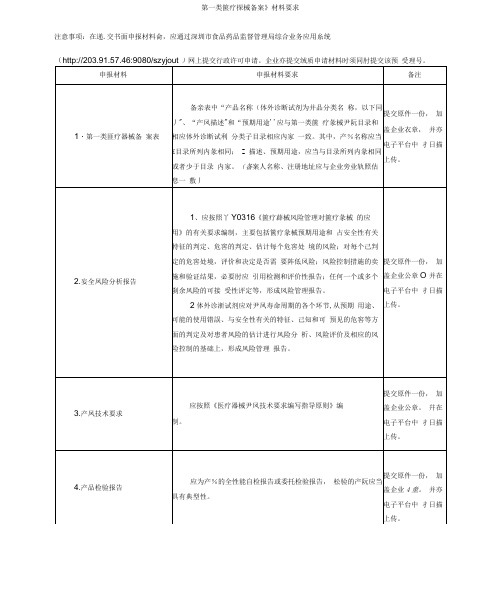
第一类箧疗探械备案》材料要求
注意事项:在递.交书面申报材料侖,应通过深圳市食品药品监督管理局综合业务应用糸统
(http://203.91.57.46:9080/szyjout )网上提交行政许可申请。
企业亦提交绒质申请材料时须同肘提交该预受理号。
I
备案资料完整齐备。
备亲表填写老整。
二、各项文件除证朗性丈件外均应以中文形式提供。
如证朗性文件为外文形式还应提供中文译本。
根据外文资料翻译的申报资料,应同时提供原丈。
b5E2RGbCAP
三、备案所有资料应由备案人签章。
“签章’、是指:备案人盖章,或者其出定代表人、负责人签名加企业盖章。
所盖章必须是备亲人公章,不得使用注册专用章。
plEanqFDPw
0、备案资料应有所提交资料目录,包括整个申报脊料的1级和2级栋题,并以表格形式说朗毎项的卷和页码。
五、材料申报人应为企业申请人或朕糸人,非上述两种人员的,应提交法定代表人或企业负责人签署的授权委托书(核
对身份证原件丿DXDiTa9E3d
六.申报材料请使用A4绒打印,或用钢笔.签字笔认真填写,并按照提交材料目录按顺序裝订成册。
第一类医疗器械备案资料要求及说明

附件1第一类医疗器械备案资料要求及说明一、备案资料(一)第一类医疗器械备案表(二)安全风险分析报告医疗器械应按照YY 0316《医疗器械风险管理对医疗器械的应用》的有关要求编制,主要包括医疗器械预期用途和与安全性有关特征的判定、危害的判定、估计每个危害处境的风险;对每个已判定的危害处境,评价和决定是否需要降低风险;风险控制措施的实施和验证结果,必要时应引用检测和评价性报告;任何一个或多个剩余风险的可接受性评定等,形成风险管理报告。
体外诊断试剂应对产品寿命周期的各个环节,从预期用途、可能的使用错误、与安全性有关的特征、已知和可预见的危害等方面的判定及对患者风险的估计进行风险分析、风险评价及相应的风险控制的基础上,形成风险管理报告。
(三)产品技术要求产品技术要求应按照《医疗器械产品技术要求编写指导原则》编制。
(四)产品检验报告产品检验报告应为产品全性能自检报告或委托检验报告,检验的产品应当具有典型性。
(五)临床评价资料1. 详述产品预期用途,包括产品所提供的功能,并可描述其适用的医疗阶段(如治疗后的监测、康复等),目标用户及其操作该产品应具备的技能/知识/培训;预期与其组合使用的器械。
2. 详述产品预期使用环境,包括该产品预期使用的地点如医院、医疗/临床实验室、救护车、家庭等,以及可能会影响其安全性和有效性的环境条件(如温度、湿度、功率、压力、移动等)。
3. 详述产品适用人群,包括目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息,以及使用过程中需要监测的参数、考虑的因素。
4. 详述产品禁忌症,如适用,应明确说明该器械禁止使用的疾病或情况。
5. 已上市同类产品临床使用情况的比对说明。
6. 同类产品不良事件情况说明。
(六)产品说明书及最小销售单元标签设计样稿医疗器械应符合相应法规规定。
进口医疗器械产品应提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
体外诊断试剂产品应按照《体外诊断试剂说明书编写指导原则》的有关要求,并参考有关技术指导原则编写产品说明书。
一类医疗器械备案申报要求及说明
产品预期用途
详述产品预期用途,包括产品所提供的功能,并可描述其适用的医疗阶段(如治疗后的监测、康复等),目标用户及其操作该产品应具备的技能/知识/培训;预期与其组合使用的器械。
产品预期使用环境
详述产品预期使用环境,包括该产品预期使用的地点如医院、医疗/临床实验室、救护车、家庭等,以及可能会影响其安全性和有效性的环境条件(如温度、湿度、功率、压力、移动等)。
对生产过程相关情况的概述。无源医疗器械应明确产品生产加工工艺,注明关键工艺和特殊工艺。有源医疗器械应提供产品生产工艺过程的描述性资料,可采用流程图的形式,是生产过程的概述。体外诊断试剂应概述主要生产工艺,包括:固相载体、显色系统等的描述及确定依据,反应体系包括样本采集及处理、样本要求、样本用量、试剂用量、反应条件、校准方法(如果需要)、质控方法等。应概述研制、生产场地的实际情况。
体外诊断试剂应对产品寿命周期的各个环节,从预期用途、可能的使用错误、与安全性有关的特征、已知和可预见的危害等方面的判定及对患者风险的估计进行风险分析、风险评价及相应的风险控制的基础上,形成风险管理报告。
3.产品技术要求
产品技术要求应按照《医疗器械产品技术要求编写指导原则》编制。
4.产品检验报告
产品检验报告应为产品全性能自检报告或委托检验报告,检验的产品应当具有典型性。
医疗器械应符合相应法规规定。进口医疗器械产品应提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
体外诊断试剂产品应按照《体外诊断试剂说明书编写指导原则》的有关要求,并参考有关技术指导原则编写产品说明书。进口体外诊断试剂产品应提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
7.生产制造信息
产品适用人群
详述产品适用人群,包括目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息,以及使用过程中需要监测的参数、考虑的因素。
第一类医疗器械生产备案资料要求(20170623版)

第一类医疗器械生产备案资料要求(版)【设定依据】1.《医疗器械监督管理条例》(国务院令第650号)2.《关于发布第一类医疗器械产品目录的通告》(国家食品药品监督管理总局通告2014年第8号)3.《关于医疗器械生产经营备案有关事宜的公告》(国家食品药品监督管理总局公告2014年第25号)4.《关于实施第一类医疗器械生产备案和第二类医疗器械经营备案有关事宜的通知》(苏食药监械管〔2014〕143号)注:苏州市政务服务中心只受理工业园区的。
姑苏区、高新区、吴中区、相城区、吴江区、昆山市、太仓市、常熟市、张家港市的生产企业请到当地县(区)局办理。
【所需材料】1.第一类医疗器械生产备案表;2.所生产产品的注册证/医疗器械备案凭证复印件(产品仅供出口的,提供拟出口第一类医疗器械产品清单);3.经核准的注册产品标准/备案的产品技术要求复印件(产品仅供出口的,提供出口第一类医疗器械产品技术要求或详细技术合同复印件);4.企业营业执照副本和组织机构代码证副本复印件,已经获得第二类、第三类医疗器械生产许可的企业申请第一类医疗器械生产备案时应递交《医疗器械生产企业许可证》复印件;5.法定代表人、企业负责人身份证明复印件;6.生产、质量和技术负责人的身份、学历职称证明复印件;7.生产场地的证明文件(有特殊生产环境要求的,还应提交设施、环境的证明文件)复印件;8.质量手册和程序文件;9.其他证明材料(如境内企业之间委托生产合同复印件,境外企业委托境内企业生产仅供出口合同复印件、质量管理体系建设承诺书、门牌号码文字性改变证明等);10.经办人授权证明和经办人身份证复印件;11.行政许可(行政确认)申请材料真实性保证声明。
12.第一类医疗器械生产备案凭证;13. 选择办理方式14.第一类医疗器械生产备案表信息表15.变更备案说明及其证明材料16.备案凭证原件17.补发备案凭证情况说明;18.备案凭证遗失后备案人向原备案部门的报告;19.备案人在原备案部门指定的媒体上登载遗失声明原件20.取消备案说明;首次备案:1-14;变更备案:1、15、16、10、11、12、13、14;补发凭证:1、17、18、19、10、11、12、13、14;取消备案:20、16、10、11。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
产品技术要求
编号:
医用帽
2020-5-10 发布2020-5-10 实施
XXXX有限公司发布
医疗器械产品技术要求编号:
医用帽
1.产品型号/规格及其划分说明
型号:/
2.性能指标
2.1医用帽的型号规格应符合表1、的规定。
表 1(医用帽)单位为厘米
帽外围直径帽口直径
尺寸30 12
偏差±2±2
2.2外观
2.2.1医用帽应干燥、清洁、无霉斑,表面不允许有粘连、裂缝、孔洞等缺陷。
2.2.2医用帽连接部位可采用针缝、粘合或热合等加工方式。
针缝的针眼应密封处理,针距每3cm 应为8针~14针,线迹应均匀、平直,不得有跳针。
粘合或热合等加工处理后的部位,应平整、
密封,无气泡。
2.3结构见结构图 1
结构图1
2.3.1医用帽的结构应合理,穿脱方便,结合部位严密。
2.3.2帽子为圆形,帽口采用弹性橡筋收口,以适应多种头型。
2.4 断裂强力医用帽关键部位材料的断裂强力应不小于45N。
注:关键部位为帽面。
2.5断裂伸长率医用帽关键部位材料的断裂伸长率应不小于15%
2.6应无异常气味。
2.7包装密封性要好,不允许漏气。
3.检验方法
3.1型号规格
使用通用量具,对医用帽样品进行测量,测定3件,其规格均应符合 2.1的要求。
3.2外观
3.2.1目视检查,应符 2.2.1的要求。
3.2.2目视检查,针距使用通用量具进行测量,应符合 2.2.2的要求。
3.3结构
目视检查,应符合 2.3的要求。
3.4断裂强力
医用帽关键部位材斜按照GB/T3923.1-1997规定的条样法进行试验,结果应符合 2.4的要求。
3.5断裂伸长率
医用帽关键部位材料按照GB/T3923.1-1997规定的条样法进行试验,结果应符合 2.5的要求。
3.6气味检验采取用鼻子闻的方法检验,符合 2.86的要求。
3.7包装密封性检验:参照GB15980标准要求,将小包装进入水中,轻轻挤压,不出气泡为合格。
符合 2.7的要求。