无菌医疗器械所需的验证和确认清单
医疗器械过程验证和确认

一、什么是过程确认(process validation)?过程确认基本上就是日常我们所说的工艺验证(process validation)。
在GHTF/SG3/N99-10:2004(第2版)中,对过程确认给出如下定义:提供客观证据,证明过程(或工艺)将能连续地生产出符合预定要求的结果或产品。
换句话说,过程确认就是在一个过程或工艺被交付用于正式或批量生产销售的产品之前,通过提供相应证据如试验结果、计算结果、对比分析等,证明该过程或工艺具备持续地生产符合要求的结果或生产合格产品能力;一般情况下,正式交付用于生产以后,还需要根据情况考虑再确认。
二、什么样的过程需要确认?关于什么样的过程需要确认,ISO13485的7.5.2.1是这样描述的:“当生产和服务提供过程的输出不能由后续的监视或测量加以验证时,组织应对任何这样的过程实施确认。
这包括仅在产品使用或服务已交付之后问题才显现的过程。
”换种方式,可以用下图判断什么样的过程需要确认(见GHTF/SG3/N99-10:2004(第2版):此处所指的“验证”,是指通过检验、试验、计算、对比分析等方法判定某过程(或工艺)的结果或产品是否合格。
其实很多过程在日常生产过程中,企业通过对其结果或产品进行全部验证然后才放行产品是不可行的,原因如下:—检验时,对产品是破坏性的;—检验活动很耗时;—检验的成本很高。
对这样的过程,因此企业应在该过程(或工艺)被交付用于正式或批量生产销售的产品之前,通过提供相应证据如试验结果、计算结果、对比分析等,证明该过程(或工艺)具备持续地生产符合要求的结果或生产合格产品能力,从此日常生产时即使不全部验证,也能判定该过程生产合格产品的能力是符合要求的。
企业如果对这样的过程不进行确认,此时应提供对该过程风险分析和风险评估的证据,且评估结果时该过程有关的风险很低,低至可以接受。
具体判定哪些过程需要确认时,可以采纳下面的流程方法:a)首先,确定生产流程,画出流程图,注意确保和实际生产流程是一致的。
医疗器械生产过程中的设备设施验证与确认

对检测设备进行定期校准和验证,确保其准确性和 可靠性。
03
采用先进的无损检测技术,提高产品检测的效率和 准确性。
产品质量控制效果的评估与改进
建立完善的质量评估机制,对产品质量 控制效果进行定期评估。
鼓励员工积极参与质量改进活动,提高 全员质量意识。
针对评估结果中存在的问题,制定相应 的改进措施并跟踪验证。
在正式生产前,对优化后的工艺流程 进行验证,确保其能够满足医疗器械 的生产要求和质量标准。
工艺流程优化
通过对生产工艺流程ቤተ መጻሕፍቲ ባይዱ分析和评估, 找出影响产品质量和生产效率的关键 因素,对工艺流程进行优化和改进, 提高生产效率和产品质量。
生产设备的布局与调整
设备布局规划
根据生产工艺流程和车间实际情况,合理规划生产设备的 布局,确保设备之间的协调性和生产流程的顺畅性。
降低产品缺陷率和不良事件发生 率
通过验证与确认,及时发现并纠正设备设 施存在的问题,降低产品缺陷率和不良事 件发生率,提高产品的安全性和可靠性。
提高生产效率和经济效益
增强企业信誉和市场竞争力
通过规范的验证与确认流程,优化生产流 程和设备配置,提高生产效率和经济效益 ,降低生产成本和浪费。
通过严格的验证与确认,确保产品质量和 安全性能得到保障,增强企业信誉和市场 竞争力,赢得消费者信任和支持。
对医疗器械生产过程进行全面梳理,找出生 产流程中的瓶颈和问题,通过改进工艺、优 化布局等措施提高生产效率。
精益生产理念引入
引入精益生产理念,通过减少浪费、提高效 率、降低成本等措施,实现医疗器械生产过 程的持续改进和优化。
THANKS
设备调整与校准
对生产设备进行调整和校准,确保其能够满足医疗器械的 生产要求和质量标准。同时,建立设备维护和保养制度, 确保设备的稳定性和可靠性。
无菌医疗器械研发生产需开展的验证和确认清单

无菌医疗器械研发生产需开展的验证和确认清单当医疗器械应当以无菌的形式提供时,在其灭菌前应将各种非预期的微生物污染降至最低。
因此,其生产场地、生产环节等均需要进行严格的无菌要求和过程控制。
为了保障无菌医疗器械的生产过程符合要求,在生产环境、质量控制等环节需进行必要的验证和确认。
本文按照无菌医疗器械生产质量管理规范等法规的具体要求,将无菌医疗器械设计开发及生产质量管控过程需开展的验证和确认项目进行归档整理,以供大家参考。
一、机构和人员(1)人员净化效果(手消毒)验证;(2)人手及物体初始菌检验方法的验证;(3)洁净工作服清洗效果验证;(4)消毒剂消毒效果验证。
二、厂房和设施(1)洁净室最大容纳人数验证;(2)洁净区(室)环境(空调净化系统)验证;(3)洁净室消毒效果验证;(3)制水(纯化水、注射用水)系统验证(含管道、储存罐清洗消毒验证);(4)压缩空气系统验证;(5)地面、墙面、顶棚清洁消毒验证;(6)人员上限验证;(7)洁净区清场有效期验证。
三、设备(1)关键生产及检测设备的验证;(2)设备、工装、工位器具清洁消毒验证;(3)检测设备(如净化工作台、生物安全柜等)验证。
四、设计开发(1)产品的设计验证;(2)产品的设计确认(吸塑封口、超声波焊接、电源线焊锡、管路涂胶、清洗预确认、清洗确认、灭菌确认);(3)设计转换的确认。
五、生产管理(1)无菌包装封口(如全塑包装、纸塑包装)过程确认;(2)产品、物料和自配试剂的有效期验证;(3)对关键过程的验证;(4)对特殊过程的确认;(5)清场及消毒的验证;(6)物料及产品清洗的验证。
六、质量控制(1)物料及产品净化效果(初始污染菌和微粒污染)验证;(2)产品初始菌和微粒污染验证及其检测方法的验证;(3)无菌检验方法验证;(4)微生物限度检验方法验证;(5)灭菌过程确认(环氧乙烷、辐照等)及使用软件的确认;(6)解析过程确认;(7)包装运输确认;(8)产品有效期验证(加速老化试验、实时老化试验)。
无菌医疗器械产品单包装验证方案

无菌医疗器械产品单包装验证方案一、适用范围:适用于我公司生产的一次性使用输液器带针、一次性使用静脉留置针(以下简称各产品)等产品的全塑单包装、吸塑单包装、PE/透析纸单包装及带方形透析纸单包装。
二、过程要求(验证项目):1、包装材料成型和密封过程的适应性2、包装完好性试验3、微生物屏障(阻菌性试验)4、包装材料灭菌过程的适应性5、包装材料与贮存过程的适应性6、变更时的再确认三、验证方案本包装是用于最终无菌医疗器械产品的包装,在规定的生产、灭菌、运输、贮存过程中,能够保持产品无菌性、完整性、有效性等特性的一次性使用包装材料。
1、验证目的:通过各种试验和过程验证来证明全塑单包装、吸塑单包装、PE/透析纸单包装及带方形透析纸单包装的包装材料能够满足预期用途。
2、适用范围:适用于我公司生产的各产品的单包装。
3.试验和验证方法及预计完成时间:1)、包装材料成型和密封过程的适应性(封口验证) 2013年1月2)、包装完好性试验(渗漏试验) 2013年1月3)、微生物屏障(阻菌性试验) 2013年1月4)、包装材料灭菌过程的适应性 2013年1月5)、包装材料与贮存过程的适应性(加速老化试验) 2013年4月6)、变更时的再确认四、验证小组人员职责权限姓名部门职位责任/权限技术部技术部经理负责制定包装验证方案,负责整个过程的技术工艺指导,编写验证报告。
生产部生产部经理负责验证方案的实施及样品的提供质管部质管部经理负责组织产品检测和包装测试技术部技术员负责整个过程的技术指导质管部检验员负责各项实验的操作质管部检验员负责各项实验的操作五、试验和过程验证一、包装材料成型和密封过程的适应性(封口验证):1、验证方案:1)目的:在规定的操作条件下对多个生产运转过程进行鉴定,来验证过程的有效性和稳定性。
2)范围:全塑单包装、吸塑单包装、PE/透析纸单包装及带方形透析纸单包装。
3)参与人员:4)验证步骤:a)连续塑料封口机。
医疗器械无菌检验方法验证方案

医疗器械无菌检验方法验证方案一、背景医疗器械无菌检验是确保医疗器械在使用前不会引起感染的关键步骤之一。
为了保证无菌检验方法的准确性和有效性,需要进行方法验证。
本文将介绍医疗器械无菌检验方法验证的方案。
二、目的本方案的目的是验证所采用的无菌检验方法对医疗器械的适用性和可靠性。
通过验证,确保该无菌检验方法能够准确判断医疗器械是否符合无菌要求。
三、验证内容1. 确定验证属性:根据实际需求,确定无菌检验方法的验证属性,例如检测灵敏度、特异性、准确性、重复性等。
2. 样品选择:选择符合验证属性需求的医疗器械样品,确保样本具有代表性。
3. 测试方案设计:根据所选验证属性,设计相应的测试方案。
包括实验组和对照组的划分、对照方法选择、实验参数设定等。
4. 实验组和对照组:按照测试方案进行实验组和对照组的划分。
实验组使用待验证的无菌检验方法,对照组使用已经验证过的或国际标准的无菌检验方法。
5. 检测过程:按照测试方案进行样品的无菌检验。
确保检测过程中的操作规范和环境条件符合要求。
6. 数据分析:将实验结果进行分析,包括验证属性的计算和对比分析等。
使用合适的统计分析方法,评估无菌检验方法的准确性和可靠性。
7. 结果评估:根据数据分析的结果,对无菌检验方法进行评估。
判断该方法是否符合验证属性的要求。
8. 结论和建议:根据评估结果,给出无菌检验方法的结论和改进建议。
如发现验证方法存在不足之处,建议进行进一步的优化和改进。
四、质量控制在验证过程中,需要进行严格的质量控制。
包括实验设备的校准、环境条件的监测、操作流程的标准化等。
同时,还需要使用合格的正、负对照进行比对,确保验证结果的准确性和可靠性。
五、参考文献[1] 国家药品监督管理局.医疗器械无菌检验方法验证指导原则[S].2015.[2] International Organization for Standardization. ISO 11137-1:2006, Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices[S].六、结语医疗器械无菌检验方法验证是确保医疗器械安全使用的重要环节。
关于无菌医疗器械生产工艺的验证和确认

关于无菌医疗器械生产工艺的验证和确认【摘要】本文结合《医疗器械生产质量管理规范无菌医疗器械实施细则和检查评定标准》法规要求,对无菌医疗器械生产工艺验证和确认过程进行分析和比较,通过纯化水系统、清洗过程的验证举例,提出了无菌医疗器械生产企业如何做好验证和确认活动的方法和建议。
【关键词】无菌产品验证确认2009年12月国家食品药品监督管理局[2009]834号文发布了《医疗器械生产质量管理规范(试行)》,及835、836号文发布了无菌、植入医疗器械的生产实施细则和检查评定标准(试行)的要求,所有无菌、植入医疗器械生产企业2011年1月1日起要执行此规范。
对生产工艺进行验证和确认,是确保无菌产品安全有效的关键环节。
要求验证和确认是规范的一个重要特征,也是企业执行规范的重点和难点。
本文结合《医疗器械生产质量管理规范无菌医疗器械实施细则和检查评定标准》要求,阐述如何开展验证、确认活动。
一验证和确认的概念《医疗器械生产质量管理规范(试行)》中对验证和确认规定了定义:验证:通过提供客观证据对规定要求已得到满足的认定;确认:通过提供客观证据对特定的预期用途或应用要求已得到满足的认定。
该定义与GB/T19000描述的基本一致。
二需要验证或确认的过程在《医疗器械生产质量管理规范无菌医疗器械实施细则和检查评定标准》中规定了相关的验证或确认的项目,归纳如下:三确认或验证的内容每个具体的过程,确认/验证的要求、方法和步骤不尽相同,但基本内容都是一致的。
主要包括:1、安装确认(IQ)对供应商所供技术资料的核查,对设备、备品备件的检查验收以及设备的安装检查,以确证其是否符合GMP、厂商的标准及企业特定技术要求的一系列活动。
2、运行确认(OQ)指通过按草拟的标准操作规程(SOP)进行单机或系统的运行试验,俗称试车。
运行确认是证明设备或系统各项技术参数能否达到设定要求的一系列活动。
3、性能确认(PQ)证明设备、系统是否达到设计标准和GMP有关要求而进行的系统性检查和试验。
《无菌、植入医疗器械实施细则》中需进行确认验证的项目
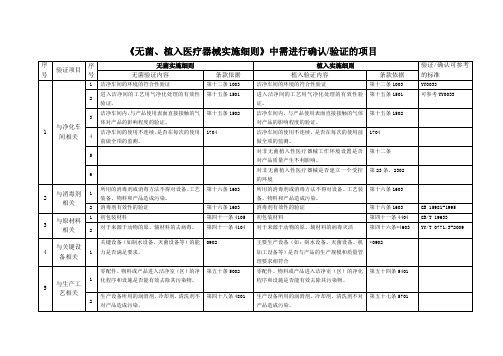
《无菌、植入医疗器械实施细则》中需进行确认/验证的项目序号验证项目序号无菌实施细则植入实施细则验证/确认可参考的标准无菌验证内容条款依据植入验证内容条款依据1 与净化车间相关1 洁净车间的环境的符合性验证第十二条1003 洁净车间的环境的符合性验证第十二条1003 YY00332进入洁净间的工艺用气净化处理的有效性验证。
第十五条1501 进入洁净间的工艺用气净化处理的有效性验证。
第十五条1501 可参考YY0033 3洁净车间内,与产品使用表面直接接触的气体对产品的影响程度的验证。
第十五条1502 洁净车间内,与产品使用表面直接接触的气体对产品的影响程度的验证。
第十五条1502 4洁净车间的使用不连续,是否在每次的使用前做全项的监测。
1704 洁净车间的使用不连续,是否在每次的使用前做全项的监测。
17045对非无菌植入性医疗器械工作环境设置是否对产品质量产生不利影响。
第十二条6对非无菌植入性医疗器械是否建立一个受控的环境第23条,23022 与消毒剂相关1所用的消毒剂或消毒方法不得对设备、工艺装备、物料和产品造成污染。
第十六条1603 所用的消毒剂或消毒方法不得对设备、工艺装备、物料和产品造成污染。
第十六条16032 消毒剂有效性的验证第十六条1603 消毒剂有效性的验证第十六条1603 GB 15981-19953 与原材料相关1 初包装材料第四十一条4105 初包装材料第四十一条4404 GB/T 196332对于来源于动物的原、辅材料的去病毒。
第四十一条4104 对于来源于动物的原、辅材料的病毒灭活第四十六条*4603 YY/T 0771.3-20094 与关键设备相关1关键设备(如制水设备、灭菌设备等)的能力是否满足要求。
0902 主要生产设备(如:制水设备、灭菌设备、机加工设备等)是否与产品的生产规模和质量管理要求相符合*09025 与生产工艺相关1零配件、物料或产品进入洁净室(区)的净化程序和设施是否能有效去除其污染物。
无菌医疗器械包装产品的要求和确认方法(2021培训课件)

EN 868 系列标准所规范的对象是以包装商品的形式在市场上流通的 产品。这些包装都是还没有形成密闭的无菌屏障系统的“半成品”, 执行标准的主体是对包装质量负责的责任人。
ISO 11607系列标准所规范的对象是已经装入器械的包装系统(无 菌屏障系统)。这时的包装不再作为独立的包装在市场上流通,而是 作为医疗器械商品的组成部分。执行标准的主体是对器械质量负责的 责任人。
4.ISO11607的要求与验证
染色渗漏试验(不透气性试验)(GB/T19633附录): 将样品包装裁成250 mm×105mm大小的样片。 取一块面积与样片相同的吸纸,放在玻璃表面,将待测试料的内表面与吸
纸接触。 将染色液倒入浅盘中,使海绵在浅盘内滞留1 min,取出海绵,靠着盘的边
把多余的液体挤除。 将海绵放在样片上,保证海绵的边缘在样片边部之内(距边部不少于
ISO11607-2《最终灭菌医疗器械的包装第2部分:成形、密封和组 装过程的确认要求》。 EN 868分为EN 868-2~-EN 868-10《待灭菌医疗器械的包装材料和
系统》系列标准。 在我国:
GB/T19633《最终灭菌医疗器械的包装 》--国家标准--ISO11607转 化而来。
YY/T0698《最终灭菌医疗器械包装材料 》--医药行业 ---EN868转 化而来。
熠品医疗与生物实验室
无菌医疗器械包装系统的 要求和确认方法
殷龙/Frank Yin
2021年07月16日
熠品上海/熠品苏州 EPIN SHANGHAI/SUZHOU LTD.
目录
1 无菌医疗器械概述 2 包装系统的组成 3 包装系统标准的现状 4 ISO11607的要求与验证 5 包装系统货架寿命确认
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
有朋友留言询问 GMP 里规定无菌医疗器械到底要做哪些验证和确认,医疗人咖啡在此做一个总 结,分享给大家。
按照 GMP 的顺序,整理如下:
机构和人员 1. 人员净化效果(手消毒验证 2. 人手及产品初始菌验证 3. 洁净工作服清洗效果验证 4. 消毒剂消毒效果验证
厂房和设施 5. 洁净室最大容纳人数验证 6. 洁净室环境验证 7. 洁净室消毒验证 8. 制水系统验证 9. 压缩空气系统验证
设备 10. 关键生产及检测设备的验证 11. 设备工装工具的清洁验证
设计开发 12. 产品的设计验证、确认及设计转换的确认
生产管理 13. 无菌包装封口过程确认 14. 产品、物料和自配试剂的有效期验证 15. 对关键过程的验证和特殊过程的确认 16. 清场及消毒的验证 17. 物料及产品清洗的验证