本地Blast
本地blast的详细用法∷柳城

本地blast的详细用法Posted on 03 四月 2009 by 柳城,阅读 9,626本地blast的详细使用方法blast all -p blastn -i myRNA.fasta -d humanRNA.fasta -o myresult.blastout -a 2 -F F -T T -e 1e-10解释如下:blastall: 这是本地化/命令行执行blast时的程序名字!(Tips:blastall直接回车就会给出你所有的参数帮助,但是英文的)-p: p 是program的简写,program在计算机领域中是程序的意思。
此参数是指定要使用何种子程序,所谓子程序,就是针对不同的需要,如核酸序列和核酸序列进行比对、蛋白质序列和蛋白质序列进行比对、假设翻译后核酸序列于蛋白质序列进行比对,选择相应的子程序: blastn 是用于核酸对核酸 blastp 是蛋白质对蛋白质序列等等,一共5个自程序。
-i: i 是input的简写,意思是输入文件,就是你自己的要进行比对的序列文件(fasta格式)-d: d是database的简写,意思是要比对的目标数据库,在例子中就是humanRNA.fasta (别忘了要formatdb)-o: o是output的简写,意思是结果文件名字,这个根据你自己的习惯起名字,可以带路径,(上边两个参数-i -d 也都可以带路径)*注意以上4个参数是必须的,缺一不可,下面的参数是为了得到更好的结果自己可调的参数,如果你不加也没有关系,blastall程序本身会给一个默认值!-a: 是指计算时要用的CPU个数,我的机器有两个CPU,所以用-a 2,这样可以并行化进行计算,提高速度,当然你的计算机就一个CPU,可以不用这个参数,系统默认值为1,就是一个CPU-F: 是filter的简写,blastall程序中有对简单的重复序列和低复杂度的一些repeats过滤调,默认是T (注意以后的有几种参数就两个选项,T/F T就是ture,真,你可以理解为打开该功能; F就是false,假,理解为关闭该功能)-T: 是HTML的简写,是指blast结果文件是否用HTML格式,默认是F!如果你想用IE看,我建议用-T T-e: 是Expectation value,期望值,默认是10,我用的10-10!BLASTALL 用法a.格式化序列数据库格式化序列数据库— —formatdbformatdb简单介绍:formatdb处理的都是格式为 ASN.1和FASTA,而且不论是核苷酸序列数据库,还是蛋白质序列数据库;不论是使用Blastall ,还是Blastpgp,Mega Blast应用程序,这一步都是不可少的。
本地BLAST

本地blast的安装及使用安装:1.首先进入NCBI2.点击ALL Resources3.点击ALL Resources里的Downloads选项卡4.点击BLAST(Stand-alone)选项在BLAST+executables中点击ftp:///blast/executables/blast+/LATEST/ . 链接(这只是说这个链接如何找到的,可以直接点击这个链接进行下载)。
5点击ncbi-blast-2.2.29+-win32.exe进行下载,大家的电脑一般为32位的,加入为64 位的则需要点击ncbi-blast-2.2.29+-win64.exe下载,根据个人情况定6下载好后点击“下一步“进行安装。
运行:1.点击电脑桌面的“开始“——”运行“,在”打开“中输入”cmd“,(这也就是调取DOS命令,快捷键”windows“+“R“键,然后回车)2切换到blast的bin目录下,例如我的路径是C:\Program Files\NCBI\blast-2.2.29+\bin,那么我的命令是:然后回车。
切换后的结果是:3把你的物种数据和比对的数据文件移动到bin文件夹下,然后做下面的。
1)建库根据你要比对的物种序列建库dos 命令:makeblastdb -in ~ -dbtype nucl/prot -out ~in 后面的‘~’里填要建库的序列文件名称,如整个水稻蛋白质组第二个‘~’里填库的名称(自己命名)nucl :建核苷酸库,prot:建蛋白质库(根据你数据要求任选一个)2)比对dos 命令:blastp/blastn -query ~ -db ~ -out ~ -evalue ~ -outfmtblastp 为比对蛋白质序列,blastn比对核苷酸序列query后面的‘~’填你要比对的序列文件名db 后面填你第一步建好库的名称out 输出最终结果名称evalue 你自己设一个期望值(5)outfmt 输出文件格式填数字6或7(1)建库结果(2)比对:结果:去bin文件夹下去寻找。
本地版BLAST程序及常用参数

值1 y -M: 所使用的打分矩阵,缺省值BLOSUM62
本地版Blast 参数(4)
y -W: 字长(Word size), 默认为0(0表示核酸为 11, 蛋白质为3)
y -S:在数据库中搜索时所使用的核酸链(strand) ,只对blastn、blastx和tblastx有效;
y 1表示top, y 2表示bottom, y 3表示both;缺省值3
本地版Blast 参数(3)
y -q: 一个核酸碱基的错配(mismatch)的罚分(只对 blastn有效),缺省值-3
y -r: 一个核酸碱基的正确匹配(match)的奖分(只对 blastn有效),缺省值1
y -b:显示的比对结果的最大数目,缺省值250 y -a: 运行BLAST程序所使用的处理器的数目,缺省
准备Database
(([swissprot-ID:HB?_*] | [swissprot-ID:HBA?_*]) | [swissprot-ID:HBB?_*])
准备查询序列
利用本地BLAST搜索黑猩猩血红蛋白
y Formatdb -i 773HB.FAS y Blastall -p blastx –d 773HB.FAS –i CHIMP.FAS –o
1. BLAST种类
本地版BLAST程序及常用参数
Formatdb
y 建库命令:formatdb y 功能:创建三个主要的文件——库索引(indices),
序列(sequences)和头(headers)文件 y 生成的文件的扩展名分别是:
y .pin、.psq、.phr(对蛋白质序列) y .nin、.nsq、.nhr(对核酸序列)
Formatdb 常用参数
如何解决本地Blast建库时出现的磁盘空间不足的问题
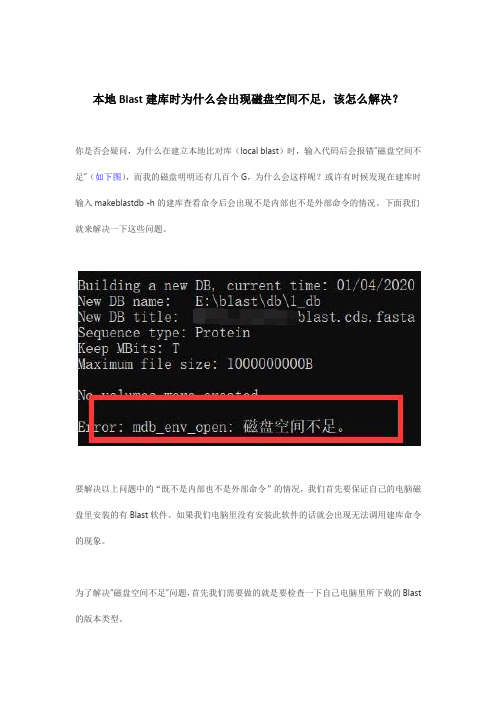
本地Blast建库时为什么会出现磁盘空间不足,该怎么解决?
你是否会疑问,为什么在建立本地比对库(local blast)时,输入代码后会报错“磁盘空间不足”(如下图),而我的磁盘明明还有几百个G,为什么会这样呢?或许有时候发现在建库时输入makeblastdb -h的建库查看命令后会出现不是内部也不是外部命令的情况。
下面我们就来解决一下这些问题。
要解决以上问题中的“既不是内部也不是外部命令”的情况,我们首先要保证自己的电脑磁盘里安装的有Blast软件。
如果我们电脑里没有安装此软件的话就会出现无法调用建库命令的现象。
为了解决“磁盘空间不足”问题,首先我们需要做的就是要检查一下自己电脑里所下载的Blast 的版本类型。
Artemis软件及本地blast工具的使用介绍

Notepad
两个文本编辑器
正则表达式
正则表达式是对字符串操作的一种逻辑公式,就是用事先定义好 的一些特定字符、及这些特定字符的组合,组成一个“规则字符 串”,这个“规则字符串”用来表达对字符串的一种过滤逻辑。
EmEditor
Artemis /science/tools/artemis
比对命令 比对类型 输入文件
数据库
-b : 显示的比对结果的最大数目,缺省值250
输出命令
E值 输出格式
缺省值10 缺省值0
-v : 单行描述(one-line description)的最大数目,缺省值500
2.3 本地blast实例分析
1. MSMEG_2092蛋白在分枝杆菌中的分布
需要文件 MSMEG_2092蛋白文件: MSMEG_2092.txt 分枝杆菌总蛋白库文件: mycobacterium_all.faa 执行命令 >formatdb -i mycobacterium_all.faa -p T >blastall -p blastp -i MSMEG_2092.txt -d mycobacterium_all.faa -o 2092out.txt -m 8 -e 1e-5 –v 1 –b 1
2. M. smegmatis 和M. tuberculosis同源基因的比对
需要文件 M. smegmatis 总蛋白文件: Ms.faa M. tuberculosis 总蛋白文件: H37Rv.faa 执行命令 >formatdb -i Ms.faa -p T >blastall -p blastp -i H37Rv.faa -d Ms.faa -o MS_H37Rv_out.txt -m 8 -e 1e-5 –v 1 –b 1
本地blast使用

蛋白质序列和核酸数据库中的核酸序列 翻译后的蛋白质序列逐一比对。
核酸序列翻译成蛋白质序列,再和核酸 数据库中的核酸序列翻译成的蛋白质 序列逐一进行比对。
(三)分析结果
示例程序: Extract_blastB.pl
1. 安装SSH 2. 登陆:内网 120.94.179.252 用户名:class 密码:123 3. 传输文件 4. 终端命令行 cd ls
一、两步完成本地BLAST:
(1) formatdb --格式化序列数据库 命令示例:formatdb -i *.fasta -p T (2)Blastall 命令示例:blastall -p blastp -i ** -d * -e 1e-4 -o ***.blast
本 地 Blast
2013-2-22 张玉娟
在 线Blast:
本 地 Blast:
(一)Windows系统单机完成; (二)上传服务器完成; (三)分析结果;
(一)Windows系统单机完成;
将需要的文件放入同一个文件夹
formatdb
blastall
产生out文件
(二)上传服务器完成;
服务器使用流程:
-m alignment view options: 比对显 示选项,其具体的说明可以用以下的比对实例说明 0 = pairwise,显示具体匹配信息(缺省) 1 = query-anchored showing identities,查询-比上区域,显示一致性 2 = query-anchored no identities,查询-比上区域,不显示一致性 3 = flat query-anchored, show identities,查询-比上区域的屏文形式,显示一致性 4 = flat query-anchored, no identities,查询-比上区域的屏文形式,不显示一致性 5 = query-anchored no identities and blunt ends, 查询-比上区域,不显示一致性,无突然的结束 6 = flat query-anchored, no identities and blunt ends, 查询-比上区域的屏文形式,不显示一致 性 7 = XML Blast output,XML格式的输出 8 = tabular,TAB格式的输出 9 =tabular with comment lines, 带注释行的TAB格式的输出 10 =ASN, text,文本方式的ASN格式输出 11 =ASN, binary [Integer] default = 0, 二进制方式的ASN格式输出
构建NCBI本地BLAST数据库(NRNT等)blastxdiamond使用方法blast。。。
构建NCBI本地BLAST数据库(NRNT等)blastxdiamond使⽤⽅法blast。
:如何下载 NCBI NR NT数据库?下载blast:先了解BLAST Databases:如何下载NCBI blast数据库?NCBI提供了⼀个⾮常智能化的脚本update_blastdb.pl来⾃动下载所有blast数据库。
脚本使⽤⽅法:perl update_blastdb.pl nr有哪些可供下载的blast数据库?perl update_blastdb.pl --showall该命令会显⽰所有可供下载的blast数据库,请⾃⾏选择:16SMicrobialcdd_deltaenv_nrenv_ntestest_humanest_mouseest_othersgssgss_annothtgshuman_genomiclandmarknrntother_genomicpataapatntpdbaapdbntref_prok_rep_genomesref_viroids_rep_genomesref_viruses_rep_genomesrefseq_genomicrefseq_proteinrefseq_rnarefseqgenestsswissprottaxdbtsa_nrtsa_ntvector这⾥我选择的是nr数据库。
nohup perl update_blastdb.pl --decompress nr >out.log 2>&1 &⾃动在后台下载,然后⾃动解压。
(下载到⼀半断⽹了,在运⾏会接着下载,⽽不会覆盖已经下载好的⽂件)blast如何使⽤?这⾥只演⽰blastx的使⽤⽅法。
刚才下载的nr库就是蛋⽩库,blastx就是⽤来将核酸序列⽐对到蛋⽩库上的。
(nt就是核酸库)因为我们下载的是已经建好索引的数据库,所以省去了makeblastdb的过程。
常见的命令有下⾯⼏个:-query <File_In> 要查询的核酸序列-db <String> 数据库名字-out <File_Out> 输出⽂件-evalue <Real> evalue阈值-outfmt <String> 输出的格式blast构建索引 | makeblastdbmakeblastdb -in mature.fa -input_type fasta -dbtype nucl -title miRBase -parse_seqids -out miRBase -logfile File_Name-in 后接输⼊⽂件,你要格式化的fasta序列-dbtype 后接序列类型,nucl为核酸,prot为蛋⽩-title 给数据库起个名,好看~~(不能⽤在后⾯搜索时-db的参数)-parse_seqids 推荐加上,现在有啥原因还没搞清楚-out 后接数据库名,⾃⼰起⼀个有意义的名字,以后blast+搜索时要⽤到的-db的参数-logfile ⽇志⽂件,如果没有默认输出到屏幕资源消耗blastx -query test.merged.transcript.fasta -db nr -out test.blastx.out其中fasta⽂件只有19938⾏。
Windows下本地blast安装方法
Windows系统下本地BLAST安装方法1.下载安装文件:以blast-2.2.23-ia32-win32.exe为例,将此安装文件放至指定目录,以G:\blast-\为例,如图所示:2. 运行安装程序:双击上述安装文件,单击运行:程序会自动在blast-文件夹下生成3个文件夹:\bin\、\data\和\doc\:3. 添加配置文件:在桌面(任意可以新建文件的地方)新建一个.txt文件,然后将其重命名为NCBI.ini,在提示更改后缀名的对话框中点是。
打开NCBI.ini,在其中写入如下两行内容:[NCBI]Data="path\data\"上边的path是你的blast安装路径,在本例中为G:\blast-,因此,NCBI.ini中的内容为:[NCBI]Data="G:\blast-\data\"写完后保存,然后将该文件复制至C:\Windows目录下:至此,本地blast-2.2.23-ia32-win32安装完毕。
4. 导入数据库:从ftp:///blast/db/上,可下载各类数据库文件,下载完毕后,将其解压至G:\blast-\data\目录下。
注意事项:1.NCBI.ini中的路径为blast所在安装路径;2.此安装办法适用与指定版本,对于blast+版本不适用,若想安装新版本,可自行到网站查阅安装办法;附:运行示例:1.打开cmd命令行;2.通过cd命令到达安装目录的bin\目录下3.通过dir命令查看全部可执行的子程序:4.使用blastall.exe进行比对:输入blastall.exe -d refseq_rna.01 -i G:\blast-\data\test_query.fa -p blastn该命令各部分的含义为:①blastall.exe:blast主程序;②-d refseq_rna.01:选择refseq_rna.01为被搜索的数据库,其数据已存至G:\blast-\data\下;③-i G:\blast-\data\test_query.fa: 选择test_query.fa为查找序列文件,注意,查找文件应输入其绝对路径,但数据库文件不用。
本地blast使用实验报告
本地blast使用实验报告本实验旨在通过使用本地blast工具对数据库中的序列进行比对分析,掌握本地blast的使用方法,并了解其在生物信息学研究中的应用。
实验材料与方法:(1)实验软件:本地blast软件(如ncbi-blast)(2)实验数据:需要进行比对分析的目标序列和数据库序列(3)实验步骤:a. 下载并安装本地blast软件。
b. 准备数据库序列和目标序列。
c. 使用blast程序对目标序列进行比对分析。
d. 获取比对结果并进行进一步分析和解释。
实验结果与分析:本地blast分析结果包括比对的得分、相似性、长度、E值等信息。
通过比对结果可以判断目标序列与数据库序列的相似程度,进一步了解目标序列在数据库中的亲缘关系与功能。
实验使用的本地blast软件可以通过指定不同的参数来调整比对的灵敏度和特异性。
一般而言,较低的E值和较高的比对得分可以表示目标序列与数据库序列的相似性较高。
另外,本地blast还可用于快速比对分析大规模的基因组和转录组数据。
通过本地blast,可以鉴定同源基因、预测新的基因家族、进行基因功能注释、分析遗传变异等。
本地blast的使用还需要注意以下几点:(1)数据库的选择:不同的数据库适用于不同的研究目的,需要根据实验的需求选择合适的数据库。
(2)参数设置:根据需要调整比对的灵敏度和特异性,以获得最佳的比对结果。
(3)结果解释:通过对比对结果的分析解释,了解目标序列的功能和进化关系。
实验结论:本地blast是一种常用的序列比对工具,通过比对目标序列与数据库序列,可以研究序列的相似性、亲缘关系和功能等。
本地blast的使用可以帮助生物信息学研究者进行基因家族预测、功能注释、遗传变异分析等研究工作,对于生物信息学的研究和应用具有重要的意义。
然而,本地blast也存在一些局限性,包括计算资源需求高、数据库维护与更新等问题。
因此,在使用本地blast进行分析时,需要根据实验的需求和条件,合理选择适当的数据库和参数设置,以获得准确、可靠的分析结果。
NCBI本地Blast 安装方法
IN HOUSE LOCAL BLAST SEARCHTo get started you need the blastall.exe and formatdb.exe (From NCBI). The rest of the perl and batch programs you might need to change the path of the directories they are pointing to or the blast option they use, could be downloaded from:/SGMD/software/blast/Blast.htmFor the programs to work withou t modifying the paths, the whole folder “Blast.zip” should be unzipped to a folder "Blast"moved under the “C:” directory.For questions or comments please contact: Imed Ben Chouikhabchouikh@I. Step one: Blasting1) Download the database that you want to blast against, for example the NT database from NCBI. If you want to use a local database, store all the sequences in a text file. The file provided by NCBI is a zipped (nt.gz) file so you have to unzip it.2) At the DOS prompt (which you can get to from windows by choosing: Start, Run, then typing: command), run formatdb.exe to create a local database from that text file or the downloaded database.Usage:formatdb –t databasename–i inputfile –p FExamples:1) formatdb –t nt–i nt –p F2)formatdb –t snc–i inputfile –p Fdatabasename is the name you want to give to your databaseinputfile is the name of the text file that contains your sequences or the name of the database that you downloaded from GenBank (technically also a text file of sequences). More about formatdb.exe information and command options can be found here:/IEB/ToolBox/C_DOC/lxr/source/doc/formatdb.txt3) Open the file BlastList.pl (using Notepad or your favorite text editor)Make the small changes as instructed in the file then save it.These are the only two changes that should be made to run the program.4) Run BlastList.pl as follows:c:> cd Blastc:\>Blast\perl BlastList.plThe file BlastList.pl automatically creates a batch file “DosBlast.bat” depending on the list of the sequence to be blasted.5) Run DosBlast.batc:\>Blast\ DosBlast.batDosBlast.bat is the actual file that does the blast search.II. Step two: Extracting data from blast results6) Move all the resulting ".txt" files to BlastOut7) Go to the directory BlastOutc:\>Blast\ cd BlastOut8) Run Hits.plc:\>Blast\BlastOut\perl Hits.plThat will move the files that returned no hits to a different directory9) Run DataExt.plc:\>Blast\BlastOut\perl DataExt.plThe output will be written to the file Blasted.txt.With Excel open (using tab delimited) the file Blasted.txt.It contains a summary of the blast results that you can save, edit, etc.LIST OF PROGRAMS:The required programs are available in this directory but here is the code for four of them in case you wish to make some modifications:1)BlastList.bat2)DosBlast.bat3)Hits.pl4)DataExt.pl============================BlastList.bat========================= #!/usr/local/bin/perl## file: BlastList.pl#### Imed Ben Chouikha#### 04/24/03#### This files creates a batch file "DosBlast.bat" based on## the list of ".seq" sequence files.## The file "DosBlast.Bat" runs separetly. (For more information see "ReadMe.doc")#### send comments to: bchouikh@# CHANGES TO MAKE# 1) Change "SCN_seq.fas" (in the first line of the program) with the name of the local# database you are Blasting against:# 2) eliminate (only if needed) anything other than the sequence files in the# "unless" statement (below, in the middle of the code).$DBNAME = "SCN_seq.fas"; ###### Replace "SCN_seq.fas" with local Database name$dirtoget="C:/Blast";opendir(IMD, $dirtoget) || die("Cannot open directory");# delete the old "DosBlast.bat" file that contains the list of sequences# to be blasted$dosfile = "DosBlast.bat";unlink($dosfile);# Get the list of the new sequence files to blast@thefiles= readdir(IMD);closedir(IMD);# Create a new file "DosBlast.bat"open(OUT,">DosBlast.bat") || die "cannot open file for writing: $!";foreach $f (@thefiles){####### Add to the list below everything other that the sequence files####### Here is the Unless statement:unless ( ($f eq ".") || ($f eq "..") || ($f eq "DosBlast.bat") || ($f eq "BlastList.pl") || ($f eq $DBNAME)||($f eq "BlastOut") || ($f eq "blastall.exe") || ($f eq "formatdb.exe")||($f eq "formatdb.log")|| ($f eq "ReadMe.doc")){@myarray = split(/\./,$f); # Old file name$extension =".txt"; # This is the new file extension@newname=@myarray[0].$extension;print(OUT "blastall -p blastn -d $DBNAME -i $f -o @newname -v 0 -b 1\n");} # end of unless} # end of foreach============================DosBlast.bat=========================# Changed this file to be automatically generated. So you do not have to worry about it# it contains lines of the form# blastall -p blastn -d $DBNAME -i $f -o @newname -v 0 -b 1# where $DBNAME is the Database name, $f and @newname are the input and output names# read from the directory by BlastList.pl=============================================================== ============================Hits.pl==============================#!/usr/local/bin/perl#### file Hits.pl#### Imed Ben Chouikha#### 04/24/03#### This files moves all the files that returned "No Hits" to the directory called "NoHits"## and deletes them from the current directory#### Send comments to: bchouikh@$dirtoget="C:/Blast/BlastOut";opendir(IMD, $dirtoget) || die("Cannot open directory");@thefiles= readdir(IMD);#closedir(IMD);## loop over the filesforeach $f (@thefiles){unless ( ($f eq ".") || ($f eq "..") || ($f eq "Blasted.txt") || ($f eq "DataExt.pl")||($f eq "NoHits") || ($f eq "Hits.pl")){open(IN, $f) || die "cannot open file for reading: $!";#open(OUT,">nohitlist.txt") || die "cannot open file for writing: $!";$nohit = "No Hits Found";$count = 0;while($lines = <IN>){chop($lines);if ($lines =~ /$nohit/){$count += 1;}else{$count = $count;}$lines += 1;} # end of while loopif ($count >= 1){close(IN);$odir="NoHits";opendir(IMD1, $odir) || die("Cannot open directory");rename($f, "$odir/$f");closedir(IMD1);#unlink($f);}else {$count = $count;}} # end of unless} # end of foreach===========================DataExt.pl============================ #!/usr/local/bin/perl#### file: DataExt.pl#### Imed Ben Chouikha#### 04/24/03#### This file extracts the E-values, best hits, and other values from the Blast result.#### send comments to: bchouikh@$dirtoget="C:/Blast/BlastOut";opendir(IMD, $dirtoget) || die("Cannot open directory");@thefiles= readdir(IMD);closedir(IMD);## loop over the filesopen(OUT,">Blasted.txt") || die "cannot open file for writing: $!";foreach $f (@thefiles){unless ( ($f eq ".") || ($f eq "..") || ($f eq "Blasted.txt") || ($f eq "DataExt.pl")||($f eq "NoHits") || ($f eq "Hits.pl")){open(IN, $f) || die "cannot open file for reading: $!";$besthit = 0;$scorecount = 0;$line0 = "";while($lines = <IN>){if ($besthit < 1) { # main while loopchop($lines);if ($lines =~ />/){$name = $lines;}elsif($lines =~ /Length =/){$length=$lines;if ($line0 =~ />/){$secondname = $secondname;}else{$secondname = $line0;}}elsif($lines =~ /Score = /){$score = $lines;}elsif($lines =~ /Identities = /){$identities = $lines;}elsif($lines =~ /Strand = /){$strand = $lines;print ( OUT "$f $name $secondname \t $length \t $score \t $identities \t $stand \n");$besthit += 1;}else{$lines = $lines;}$line0 = $lines;$lines +=1;}else{$lines +=1;}} # end of while loop} # end of unless} # end of foreach。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
本地Blast使用说明
一、软件的下载安装
1.1安装流程
建议安装在非系统盘,如将下载的 BLAST 程序安装到 E:\blast,生成bin、doc 两个子目录,其中 bin 是程序目录,doc 是文档目录,这样就安装完毕了。
1.2 设置环境变量
右键点击“我的电脑”-“属性”,然后选择“高级系统设置”标签-“环境变量”(图1),在用户变量下方“Path”随安装过程已自动添加其变量值,即“E:\Blast\bin”。
此时点击“新建”-变量名“BLASTDB”,变量值为“E:\Blast\db”(即数据库路径,图2)。
二、查看程序版本信息
点击 Windows 的“开始”菜单下的“运行”,输入“cmd”调出 MS-DOS 命令行,转到 Blast 安装目录,输入命令“blastn -version”即可查看版本,若能显示说明本地blast 已经安装成功。
三、使用
3.1本地数据库的构建
下载所需的数据(Fasta格式),将X 放到E:\blast\db 文件夹下,然后调出MS-DOS 命令行,转到E:\blast\db 文件夹下运行以下命令:格式化
数据库,命令为:
makeblastdb -in 数据库文件 -dbtype 序列类型(核酸:nul;蛋白:prot)-title database_title-parse_seqids -out database_name-logfile File_Name
格式化数据库后,创建三个主要的文件——库索引(indices),序列(sequences)和头(headers)文件。
生成的文件的扩展名分别是:.pin、.psq、.phr(对蛋白质序列)或.nin、.nsq、.nhr(对核酸序列)。
而其他的序列识别符和索引则包含在.psi和.psd(或.nsi 和.nsd)中。
3.2核酸序列相似性搜索
blastn -db database_name -query input_file -out output_file
-outfmt "7 qacc sacc qstart qend sstart send length bitscore evalue pident ppos"
备注:qacc:查询序列Acession号;sacc:目标序列Acession号;
qstart qend:分别表示查询序列比对上的起始、终止位置;
sstart send:分别表示目标序列比对上的起始、终止位置;
length:长度; bitscore:得分; evalue:E-Value值;
pident:一致性; ppos:相似性
3.3 查看并获取目标序列:
blastdbcmd -db refseq_rna -entry 224071016 -out test.fa
可以从数据库中提取gi号为224071016的序列,并且以fasta格式存入文
件
3.4蛋白质序列相似性搜索
Blastp -db database_name-query input_file -out output_file
-outfmt "7 qacc sacc qstart qend sstart send length bitscore evalue pident ppos"
3.5 查看并获取目标序列:重复3.3。