Blast本地化详细流程
本地Blast

本地Blast使用说明一、软件的下载安装1.1安装流程建议安装在非系统盘,如将下载的 BLAST 程序安装到 E:\blast,生成bin、doc 两个子目录,其中 bin 是程序目录,doc 是文档目录,这样就安装完毕了。
1.2 设置环境变量右键点击“我的电脑”-“属性”,然后选择“高级系统设置”标签-“环境变量”(图1),在用户变量下方“Path”随安装过程已自动添加其变量值,即“E:\Blast\bin”。
此时点击“新建”-变量名“BLASTDB”,变量值为“E:\Blast\db”(即数据库路径,图2)。
二、查看程序版本信息点击 Windows 的“开始”菜单下的“运行”,输入“cmd”调出 MS-DOS 命令行,转到 Blast 安装目录,输入命令“blastn -version”即可查看版本,若能显示说明本地blast 已经安装成功。
三、使用3.1本地数据库的构建下载所需的数据(Fasta格式),将X 放到E:\blast\db 文件夹下,然后调出MS-DOS 命令行,转到E:\blast\db 文件夹下运行以下命令:格式化数据库,命令为:makeblastdb -in 数据库文件 -dbtype 序列类型(核酸:nul;蛋白:prot)-title database_title-parse_seqids -out database_name-logfile File_Name格式化数据库后,创建三个主要的文件——库索引(indices),序列(sequences)和头(headers)文件。
生成的文件的扩展名分别是:.pin、.psq、.phr(对蛋白质序列)或.nin、.nsq、.nhr(对核酸序列)。
而其他的序列识别符和索引则包含在.psi和.psd(或.nsi 和.nsd)中。
3.2核酸序列相似性搜索blastn -db database_name -query input_file -out output_file-outfmt "7 qacc sacc qstart qend sstart send length bitscore evalue pident ppos"备注:qacc:查询序列Acession号;sacc:目标序列Acession号;qstart qend:分别表示查询序列比对上的起始、终止位置;sstart send:分别表示目标序列比对上的起始、终止位置;length:长度; bitscore:得分; evalue:E-Value值;pident:一致性; ppos:相似性3.3 查看并获取目标序列:blastdbcmd -db refseq_rna -entry 224071016 -out test.fa可以从数据库中提取gi号为224071016的序列,并且以fasta格式存入文件3.4蛋白质序列相似性搜索Blastp -db database_name-query input_file -out output_file-outfmt "7 qacc sacc qstart qend sstart send length bitscore evalue pident ppos"3.5 查看并获取目标序列:重复3.3。
BLAST使用方法

BLAST使用方法BLAST(Basic Local Alignment Search Tool)是一种用于比较生物学序列的工具,可以在数据库中查找类似序列,并计算它们之间的相似度。
BLAST可用于寻找相似的基因、蛋白质序列、DNA序列等,以及用于确定序列的功能和进化关系。
本文将介绍BLAST的使用方法。
2. 准备序列:在使用BLAST之前,你需要准备你想要比较的序列。
可以是DNA序列、蛋白质序列或其他生物学序列。
可以从公共数据库如NCBI的GenBank中获取序列,也可以使用你自己的实验数据。
3.选择数据库:BLAST使用数据库来存储和检索序列。
常见的数据库包括NCBI的NT数据库(核苷酸数据库),NR数据库(非冗余蛋白质数据库)等。
根据你的研究需要,选择适合你的数据库。
你也可以建立自己的数据库,将实验室内部的数据添加到其中。
4.运行BLAST:使用BLAST的命令行接口或网页界面,输入你的序列和数据库信息,运行BLAST。
下面是使用命令行接口运行BLAST的示例:`$ blastn -query sequence.fasta -db nt -out result.txt`在这个命令中,`blastn`是BLAST程序的名称,`sequence.fasta`是包含你的序列的FASTA文件,`nt`是数据库的名称,`result.txt`是结果输出的文件。
如果使用网页版BLAST,你只需将序列和数据库信息输入网页表单,点击运行即可。
5.解析结果:BLAST运行完成后,会生成一个结果文件,其中包含比对结果和相似度分数。
你可以使用BLAST提供的工具来解析和可视化这些结果,以便进一步分析。
结果中通常包括比对的相似度分数、比对的位点、比对的长度、匹配的碱基或氨基酸序列等。
通过分析结果,你可以确定序列的功能和进化关系,或者寻找可能的同源序列。
6.参数调整:BLAST提供了许多参数用于调整比对过程和结果的特性。
Blast本地化安装图解

Blast本地化:window平台下blast软件的安装boyun发表于 2009-07-09 17:08 | 阅读 1 views1.对于windows 2000/xp 用户,下载blast-2.2.18-ia32-win32.exe安装文件ftp:///blast/executables/LATEST/blast-2.2.18-ia32-win32.exe2.创建一个新目录,例如C:\blast,将下载的文件blast-2.2.18-ia32-win32.exe复制到该目录,双击这个文件,自解压产生bin、data、doc 三个目录,bin是程序目录,data是程序使用数据的目录,doc是文档目录。
表:bin目录中的程序程序说明bl2seq.exe进行两条序列比对blastall.exe做普通的blast比对blastclust.exeblastpgp.execopymat.exefastacmd.exe通过gi号,接收号等,在数据库中检索序列formatdb.exe格式化数据库formatrpsdb.exeimpala.exemakemat.exemegablast.exe megablast程序rpsblast.exeseedtop.exe3.用文本编辑器创建一个ncbi.ini文件,文件包含下面内容:[NCBI]Data="C:\blast\data\"将ncbi.ini文件存放到系统的Windows 或者 WINNT目录。
4.将”C:\blast\bin”目录添加路径中(该步骤非必须,但会给以后的操作带来方便),方法:1)右击我的电脑选择属性,选择高级,点击环境变量,设置环境变量2)系统变量中,选择Path,点击“编辑”,在变量值的后面添加“; C:\blast\bin”,点击确定将安装路径添加到path5.测试,打开dos窗口(点击开始,选择运行,打开的输入框中输入“cmd”,确定),键入“blastall”,回车,如果安装正确,将显示blastall的所有参数说明。
如何本地化进行blast序列比对

如何本地化进行blast序列比对1、基本概念相似性(Similarity)是指序列比对过程中用来描述检测序列和目标序列之间相同或相似碱基或氨基酸残基占全部比对碱基或氨基酸残基的比例的高低,属于量的判断。
同源性(Homology)是指从某一共同祖先经趋异进化而形成的不同序列。
只有当两个蛋白质在进化关系上具有共同的祖先时,才可称它们为同源的,属于质的判断。
相似性和同源性的关系当相似程度高于50%时,比较容易推测检测序列和目标序列可能是同源序列;而当相似性程度低于20%时,就难以确定或者根本无法确定其是否具有同源性。
总之不能把相似性和同源性混为一谈。
所谓“具有50%同源性”,或“这些序列高度同源”等说法,都是不确切的,应避免使用。
序列相似性比较和同源性分析序列相似性分析:就是用来计算待研究序列与某序列之间的相似性程度,常用的软件包有BLAST、FASTA等;序列同源性分析:是将待研究与来自不同物种的序列中进行进化分析,以确定该序列与其它序列间的亲源关系。
常用的程序包有Phylip及Mega等进化分析软件;全局比对与局部比对全局比对寻找序列在全长范围内最佳比对。
常用算法如:Needleman-Wunsch algorithm(Needle)在线程序如: Needle局部比对寻找序列在局部区域的最高比对打分。
常用算法如:Smith-Waterman algorithm, blast,fasta等在线程序如: WaterNeedle及Water的在线程序也可以本地安装Emboss执行以上程序局部相似性比对的生物学基础蛋白质功能位点往往是由较短的序列片段组成的,尽管在序列的其它部位可能有插入、删除等突变,但这些关键的功能部位的序列往往具有相当大的保守性。
而局部比对往往比整体比对对这些功能区段具有更高的灵敏度,因此其结果更具生物学意义。
BLAST程序常用的两个评价指标Score:使用打分矩阵对匹配的片段进行打分,这是对各对氨基酸残基(或碱基)打分求和的结果,一般来说,匹配片段越长、相似性越高则Score值越大,结果越可信。
Windows下本地blast安装方法
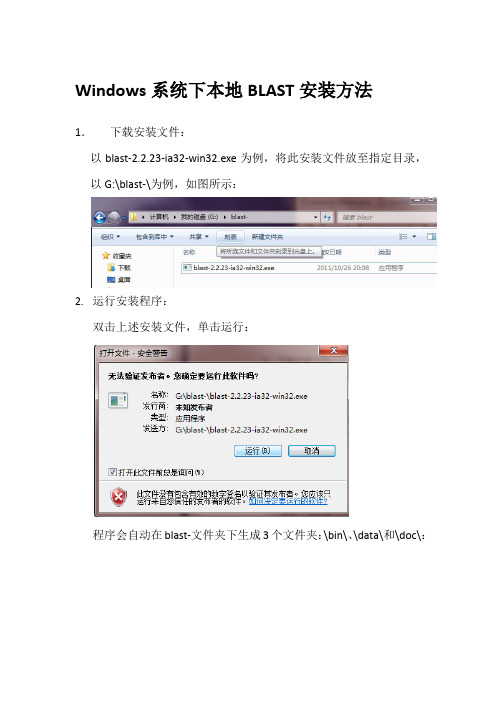
Windows系统下本地BLAST安装方法1.下载安装文件:以blast-2.2.23-ia32-win32.exe为例,将此安装文件放至指定目录,以G:\blast-\为例,如图所示:2. 运行安装程序:双击上述安装文件,单击运行:程序会自动在blast-文件夹下生成3个文件夹:\bin\、\data\和\doc\:3. 添加配置文件:在桌面(任意可以新建文件的地方)新建一个.txt文件,然后将其重命名为NCBI.ini,在提示更改后缀名的对话框中点是。
打开NCBI.ini,在其中写入如下两行内容:[NCBI]Data="path\data\"上边的path是你的blast安装路径,在本例中为G:\blast-,因此,NCBI.ini中的内容为:[NCBI]Data="G:\blast-\data\"写完后保存,然后将该文件复制至C:\Windows目录下:至此,本地blast-2.2.23-ia32-win32安装完毕。
4. 导入数据库:从ftp:///blast/db/上,可下载各类数据库文件,下载完毕后,将其解压至G:\blast-\data\目录下。
注意事项:1.NCBI.ini中的路径为blast所在安装路径;2.此安装办法适用与指定版本,对于blast+版本不适用,若想安装新版本,可自行到网站查阅安装办法;附:运行示例:1.打开cmd命令行;2.通过cd命令到达安装目录的bin\目录下3.通过dir命令查看全部可执行的子程序:4.使用blastall.exe进行比对:输入blastall.exe -d refseq_rna.01 -i G:\blast-\data\test_query.fa -p blastn该命令各部分的含义为:①blastall.exe:blast主程序;②-d refseq_rna.01:选择refseq_rna.01为被搜索的数据库,其数据已存至G:\blast-\data\下;③-i G:\blast-\data\test_query.fa: 选择test_query.fa为查找序列文件,注意,查找文件应输入其绝对路径,但数据库文件不用。
Blast软件的详细使用方法

Blast软件的详细使用方法blastall -p blastn -i myRNA.fasta -d humanRNA.fasta -o myresult.blastout -a 2 -F F -T T -e 1e-10解释如下:blastall: 这是本地化/命令行执行blast时的程序名字!(Tips:blastall直接回车就会给出你所有的参数帮助,但是英文的)-p: p 是program的简写,program在计算机领域中是程序的意思。
此参数是指定要使用何种子程序,所谓子程序,就是针对不同的需要,如核酸序列和核酸序列进行比对、蛋白质序列和蛋白质序列进行比对、假设翻译后核酸序列于蛋白质序列进行比对,选择相应的子程序: blastn 是用于核酸对核酸blastp 是蛋白质对蛋白质序列等等,一共5个自程序。
-i: i 是input的简写,意思是输入文件,就是你自己的要进行比对的序列文件(fasta格式)-d: d是database的简写,意思是要比对的目标数据库,在例子中就是humanRNA.fasta (别忘了要formatdb)-o: o是output的简写,意思是结果文件名字,这个根据你自己的习惯起名字,可以带路径,(上边两个参数-i -d 也都可以带路径)*注意以上4个参数是必须的,缺一不可,下面的参数是为了得到更好的结果自己可调的参数,如果你不加也没有关系,blastall程序本身会给一个默认值!-a: 是指计算时要用的CPU个数,我的机器有两个CPU,所以用-a 2,这样可以并行化进行计算,提高速度,当然你的计算机就一个CPU,可以不用这个参数,系统默认值为1,就是一个CPU-F: 是filter的简写,blastall程序中有对简单的重复序列和低复杂度的一些repeats过滤调,默认是T (注意以后的有几种参数就两个选项,T/F T就是ture,真,你可以理解为打开该功能; F就是false,假,理解为关闭该功能)-T: 是HTML的简写,是指blast结果文件是否用HTML格式,默认是F!如果你想用IE看,我建议用-T T-e: 是Expectation value,期望值,默认是10,我用的10-10!BLASTALL 用法a.格式化序列数据库格式化序列数据库——formatdbformatdb简单介绍:formatdb处理的都是格式为ASN.1和FASTA,而且不论是核苷酸序列数据库,还是蛋白质序列数据库;不论是使用Blastall ,还是Blastpgp,Mega Blast应用程序,这一步都是不可少的。
Local BLAST简明教程
2.在DOS系统中利用makeblastdb.exe构建数据库
命令行:
$ makeblastdb.exe –in protein.fasta –out protein_db –hash_index–dbtype prot
构建数据库
“开始”->“运行”(快捷键Windows键+R)->输入“cmd”,“确认”->进入DOS系统 1.打开数据库(即序列文件)所在文件夹;2. 输入命令对数据库进行格式化。
打开数据库所在文件夹 运行命令,对数据库进行格式化
格式化成功
构建数据库
方法二 从NCBI中的ftp库下载所需要的数据库, 地址:ftp:///blast/db
如蛋白序列比对
$ blastp.exe –query proteins.fasta –db all_proteins_db –out blast_result.xls
待比对序列名称,
数据库名称,
结果文件名称,不同后
需要后缀
不需要后缀
缀可输出不同格式的文
件,xls为excel格式,txt、
xml等均可
要格式化的序列 的名称,需要后 缀
输出的数据库 的名称,不需 要后缀
输出的数据库 的类型 核酸:nucl 蛋白:prot
构建数据库
如:下载Aspergillus ruber基因组蛋白氨基酸序列,构建数据库 首先在NCBI上检索该物种基因组
构建数据库
基因组内共有10066个蛋白
构建数据库
LOCAL BLAST 简明教程
本地BLAST操作
本地BLAST
搜索符合条件的序列,在结果列表页面中,点击右侧Send to的下拉菜单,选择File,选择FASTA,然后产生文件保存到自己磁盘:
安装blast本地软件,并测试,点击开始菜单,选择运行,在打开的输入框中输入“cmd”,确定,进入DOS界面,路径切换到你的blast安装目录bin目录下,键入“blastall”,回车:
运行建库程序formatdb:
建库的过程是建立目标序列的索引文件,前面下载保存的fasta格式的序列文件必须用formatdb格式化后,才能用于本地BLAST搜索。
在D:\blast\bin下新建文件夹database, 将local blast demo data解压到本地,将其中的db. seq和query. fasta复制到database文件夹,在DOS窗口切换到当前目录下(d:\blast\bin),输入命令:formatdb.exe –i database\db.seq –p F
此时database文件夹下生成几个新的文件。
扩展名为nin, nsq和nhr的三个文件分别是新建的库索引(indices)、序列(sequences)和头(header)文件。
如果你的数据库是蛋白质序列,这三个文件的扩展名将分别是*.pin, *.psq和*.phr:
输入如下命令:
blastall.exe –p blastn -i database\query.fasta -d database\db.seq -o out.txt
运行结束,在bin文件夹下出现一个新文件out.txt:
附加题:(由于电脑配置问题,线上blast只做了前十条序列)。
本地BLAST使用方法
formatdb -i month.nt -p F -o T
-i input file 参数用于指定需要格式的数据库
-p type of file 用于指定文件类型,T 为蛋白质,F为核酸,默认为 T
-o parse options 用于指定是否解析序列ID并创建索引 T 为创建,F为不创建,默认为F。
blastall -p blastn -d month.nt -i test.txt -o out.txt –m 8 –e 10e-20
-p program name 为需要使用的程序名
blastn 为核酸序列对比搜索
blastp 为蛋白质序列对比搜索
blastx 为用被翻译的核酸序列在蛋白质数据库中搜索
tblastn 为用蛋白质序列在 [核酸序列翻译后数据库] [**1]中搜索
tblastx 为用翻译后的核酸序列在核酸序列翻译后数据库中搜索
-d database name 指定所使用的数据库名称
-i input file 待搜索的序列文件
-o output file 指定保存结果的文件
-m 8 表格形式输出
-e 10e-20 设置e value的值
即可在out.txt中得到相应的结果。
此外,之前由于在使用formatdb.exe 使没有使用 -o T 参数,导致没有生成索引文件,出现了以下错误提示:
WARNING: Test: Could not find index files for database month.nt
一个正确的解决办法,那就是在使用formatdb.exe时,不要忘了-o 参数,因为这个参数默认是不创建索引的,另外数据库的类型不要弄错了!。
Windows系统下本地blast
TSA:
Transcriptome Shotgun Assembly
blastn -help
blastn –query sesametest.txt –db sesameESTdb –out 002.txt
打开 002.txt 文件,查看本次blastn 结果。
对sesametest.txt 运行blastn 搜索 sesameNTdb 数据库
指一种很直接的数量关系,部分相同或
相似的百分比或其它一些合适的度量。
比如,A序列和B序列的相似性是80%, 这是个量化的关系,可进行局部比较。
3
生物序列的同源性(不可量化)
同源性(homology):
指从一些数据中推断出的两个基因或蛋白质
序列具而共同祖先的结论,属于质的判断。
即在A和B的关系上,只有是同源序列,或
3.查询序列可能是某功能基因的同源基因 „
9
两种版本的Blast比较(一)
网络版本 包括NCBI在内很多网站提供在线blast服务。
优点:使用方便,容易操作,数据库同步更新。 缺点:不利于操作大批量数据,不能构建自己的 数据库。
10
两种版本的Blast比较(二)
单机版
通过NCBI的ftp站点获得,有适合不同平台的版本(包 括linux,win,dos等)。
开发的一个基于序列相似性的数据库搜索程序。
BLAST是“基本局部对比搜索工具” (Basic Local Alignment Search Tool)的缩写。
BLAST是一套在蛋白质数据库或DNA数据库中进行 相似性比较的分析工具。
2
生物序列的相似性(可量化)
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Blast 2.4.0+本地化详细流程(基于Windows系统)1.程序获得。
从NCBI上下载Blast本地化程序,下载地址:ftp:///blast/executables/blast+/LATEST/64×安装版▲64×解压(绿色)版▲最好安装或解压到X盘根目录:如X:\blast,尽量简短,方便后边命令输入。
2.原始序列获得。
方法1:找到转录组测序数据unigene数据库文件:unigene.fasta或unigene.fa,若为unigene.fa则直接改后缀为.fasta即可。
找到或修改后将数据库文件移动至Blast本地化程序目录“X:\blast\bin”。
方法2:从NCBI中的ftp 库下载所需要库,链ftp:///blast/db/FASTA/,其中nr.gz为非冗余的数据库,nt.gz为核酸数据库,month.nt.gz为最近一个月的核酸序列数据。
下载的month.nt.gz先用WINRAR解压缩,然后用makeblastdb.exe格式化。
方法3:利用新版blast自带的update_blastdb.pl进行下载,这需要安装perl程序。
注释:上述三种方法各有优缺点,前两种下载速度较快,但是每次进行检索都需要对数据库进行格式化(转化成二进制数据),第三种方法下载速度较慢,但是NCBI 中已经格式化好的,在进行本地检索时不需再进行格式化,直接用即可。
3.用文本编辑器(txt文件改名字及后缀)创建一个ncbi.ini文件,文件包含下面内容:[NCBI]Data="C:\blast\data\" 先新建TXT文件,然后改属性,将ncbi.ini文件存放到C:\Windows4.将Blast本地化程序目录添加路径中(该步骤非必须,但会给以后的操作带来方便),方法:a)右击我的电脑选择属性,选择高级,点击环境变量,设置环境变量b)系统变量中,选择Path,点击“编辑”,在变量值的后面添加Blast本地化程序所在路径,E:\blast 点击确定,将安装路径添加到path。
5.运行MS-DOC。
打开DOC窗口(点击开始,选择运行,打开的输入框中输入“CMD”,确定),访问Blast本地化程序所在文件夹,依次输入:(1)X: 回车;(2)cd blast\bin,回车。
6.数据初始化。
下载得到的数据库为fasta格式,需要经过格式转化才能建立本地数据库。
上接第5(2)步,回车后,输入格式化数据库命令:(右键可粘贴)makeblastdb.exe –in xxx.fasta -parse_seqids -hash_index -dbtype prot,回车,在原数据库文件所在文件夹生成一系列文件,Blast本地化体系构建完成。
blast本地化命令▲blast本地化后生成的文件▲参数注释:-in参数后面接将要格式化的数据库;-parse_seqids,-hash_index两个参数一般都带上,主要是为blastdbcmd取子序列时使用,-dbtype后接所格式化的序列的类型,核酸用nucl,蛋白质用prot。
7.待比对文件建立。
在blast\bin文件夹创建test.txt文件,将需要blast的序列以fasta格式存于该文件中,文件名自己命名即可,这里以test为例。
建立fasta 文件注意事项请查看附件1。
若有NCBI上下载好的.fasta文件,直接放到blast\bin文件夹即可。
test.fasta格式文件制作▲8.本地Blast比对。
上接第6步,在MS-DOS窗口输入比对命令:blastn.exe -taskblastn -query test.fasta -db xxx.fasta -out text.txt,稍等片刻,Blast结果即存于系统自动生成的out.txt文件中。
blastn.exe -task blastn -query RefGene.txt -db Stellera.Unigene.fasta -out RefGene(test).txt -evalue 1e-5 -num_threads 8参数注释:blastn.exe为程序执行命令,程序根据自己需要而blastn,blatp,tblastx;-task后面选择你所要用的程序blastn,blatp,tblastx等;-query 后接查询序列的文件名称;-db后接格式化好的数据库名称;-out 后接输出的文件名称及格式。
by malapidan2016.08.24附件1FASTA格式说明1.构建FASTA格式文件所有TEST序列输入必须是FASTA格式,所谓FASTA是指DNA 序列第一行开始于一个标识符:">",紧接着(没有空格)是对该序列的唯一描述(即ID),然后一个空格,接着是对该序列的描述(也可以没有),从第二行开始就是一行行的序列,中间的空格,换行没有影响。
为了方便阅读,每一行序列最好不要超过80个字母。
下面是FASTA格式的示例:>Mus_AQP11 mRNA for aquaporin 11, complete cds GCGGTGAGGGAGCCATGTCCGCGCTACTGGGACTCCGGCCCGAGGTGCAG GACACCTGCATCTCGCTGGGGCTAATGCTG CTGTTCGTGCTGTTCGTGGGGCTGGCCCGCGTGATCGCCCGGCAACAGCT ACACAGGCCCGTGGTCCACGCCTTCGTCCT GGAGTTTCTAGCTACCTTCCAGCTCTGCTGCTGCACCCACGAGCTCCAAGT GCTGAGCGAGCAGGACTCTGCGCACCCCA CCTGGACTCTGACACTGATCTACTTCTTTTCCTTGGTGCATGGCCTGACCC TGGTGGGCACAGCTAGCAACCCGTGCGGC GTGATGATGCAGATGATTCTGGGGGGTATGTCCCCCGAAATGGGTGCCGT GAGGTTGTTGGCTCAGCTGGTTAGCGCCCT GTGCAGCAGGTACTGCATAAGCGCCCTGTGGAGCCTGAGTCTGACCAAGT ACCATTACGACGAAAGGATCTTAGCTTGCA GGAATCCCATCCACACCGACATGTCCAAAGCGATCATCATAGAGGCCATC TGCTCCTTTATTTTCCACAGCGCTCTACTG CACTTCCAGGAGGTCCGAACCAAGCTTCGCATCCACCTGCTGGCTGCACT CATCACCTTTTTGGCCTATGCAGGAGGGAG CCTCACAGGAGCATTGTTTAACCCAGCGCTGGCACTTTCTCTGCACTTTCC GTGCTTTGACGAACTCTTCTATAAGTTTT TTGTAGTATACTGGCTTGCTCCTTCTGTAGGTGTGCTGATGATGATCCTCA TGTTCAGTTTTTTCCTTCCATGGCTGCAT AACAATCAAATGACTAATAAAAAAGAGTAACCACTCCCAAAGACTCGAA CTAAGTCCCAGGACAGTCAAGCTGGATGCGA CAATCTGAGCACCCTCCAAACTCTGGACGCCTCCTGCTTCAGCTTTCTCTG TGGAA> Mus_AQP12 mRNA for aquaporin 12, complete cdsCCGGGCCCCCCCTCGATTGGCCAAATCGGCCCTCGAGTTAATTAAATTAAT CCCCCCCCCCCCCGTTGGGCTGTGGGACC AGCCAGTCTCCCACACGTCACCAGGTCCTTGCTCCTTGTAGAACCCAGACT GATGGCCAGTCTGAATGTGTCCCTCTGTT TCTTTTTTGCTACTTGTGCCATCTGTGAGGTGGCTAGAAGGGCATCTAAAG CCCTGCTTCCAGCAGGTACCTATGCCAGT TTTGCCCGGGGGGCAGTAGGCGCAGCCCAGCTGGCAGCCTGCTGCCTGGA GATGCGAGTGTTGGTGGAGCTTGGCCCCTG GGCAGGGGGCTTCGGACCCGACCTGTTGCTGACCCTGGTCTTCCTGCTTTT CCTGGTACATGGGGTCACCTTCGATGGGG CCTCTGCCAACCCCACCGTGGCCCTGCAGGAGTTCCTCATGGTGGAGGCA TCGCTGCCCAACACTCTGCTGAAACTGTCG GCCCAGGTGCTGGGTGCACAGGCTGCCTGTGCCCTGACCCAGCGCTGCTG GGCCTGGGAGCTCAGCGAACTACACTTACT ACAGAGCCTCATGGCTGCACACTGCAGCTCAACCCTGCGTACATCCGTGC TGCAGGGCATGCTCGTGGAGGGTGCCTGCA CCTTCTTCTTCCATCTGAGCCTCCTCCACCTGCAGCACAGCCTTCTTGTCTA CAGGGTGCCTGCCCTGGCCCTGCTGGTC ACTCTCATGGCCTACACAGCAGGGCCCTACACATCTGCCTTCTTCAATCCT GCCCTGGCTGCCTCTGTCACATTCCACTG CCCTGGGAACACCTTGCTGGAGTATGCCCACGTGTACTGCCTGGGTCCTGT CGCAGGGATGATCCTGGCTGTCCTCCTCC ATCAGGGCCACCTTCCCCGCCTTTTCCAGAGAAATCTGTTCTACCGGCAGA AAAGCAAATACCGAACTCCCAGGGGGAAG CTGTCCCCAGGTTCTGTGGACGCCAAGATGCACAAAGGGGAGTAGTGGCA AAGGGCCGTGCCCTACAGGTGCCAGGGCAG CAGCCACTGGGGTCCAGCTGCGCTGTCTCACTCACCGCAGCTTCACTCGCC TCCTGAGAGGTCTGGTCTCCCTGCCACAA AATCATTTGCCAATAAACCACTGTTAAGATCAAAAAAAAAAAAAAAAGA GCTCGGCCATAAGGGCCATAGCTCCAGCTTT TGTTCCCTTTAGTGAGGGTTAATTTCCGAGCTTGGCGTAATCA2. 从NCBI下载FASTA格式的核酸序列。