色谱图出图要求
液相色谱的结构原理和基本知识 液相色谱操作规程

液相色谱的结构原理和基本知识液相色谱操作规程液相色谱(HPLC)法是以高压下的液体为流动相,并采用颗粒极细的固定相的柱色谱分离技术。
液相色谱对样品的适用性广,不受分析对象挥发性和热稳定性的限制,因而弥补了气相色谱法的不足。
在目前已知的有机化合物中,可用气相色谱分析的约占20%,而80%则需用液相色谱来分析。
液相色谱和气相色谱在基本理论方面没有显著不同,它们之间的重大差别在于作为流动相的液体与气体之间的性质的差别。
液相色谱分析原理(1)、液相色谱分析的流程:由泵将储液瓶中的溶剂吸入色谱系统,然后输出,经流量与压力测量之后,导入进样器。
被测物由进样器注入,并随流动相通过色谱柱,在柱上进行分离后进入检测器,检测信号由数据处理设备采集与处理,并记录色谱图。
废液流入废液瓶。
遇到复杂的混合物分离(极性范围比较宽)还可用梯度控制器作梯度洗脱。
这和气相色谱的程序升温类似,不同的是气相色谱改变温度,而HPLC改变的是流动相极性,使样品各组分在佳条件下得以分离。
(2)、液相色谱的分离过程:同其他色谱过程一样,HPLC也是溶质在固定相和流动相之间进行的一种连续多次交换过程。
它借溶质在两相间分配系数、亲和力、吸附力或分子大小不同而引起的排阻作用的差别使不同溶质得以分离。
开始样品加在柱头上,假设样品中含有3个组分,A、B和C,随流动相一起进入色谱柱,开始在固定相和流动相之间进行分配。
分配系数小的组分A不易被固定相阻留,较早地流出色谱柱。
分配系数大的组分C在固定相上滞留时间长,较晚流出色谱柱。
组分B的分配系数介于A,C之间,第二个流出色谱柱。
若一个含有多个组分的混合物进入系统,则混合物中各组分按其在两相间分配系数的不同先后流出色谱柱,达到分离之目的。
不同组分在色谱过程中的分离情况,首先取决于各组分在两相间的分配系数、吸附能力、亲和力等是否有差异,这是热力学平衡问题,也是分离的首要条件。
其次,当不同组分在色谱柱中运动时,谱带随柱长展宽,分离情况与两相之间的扩散系数、固定相粒度的大小、柱的填充情况以及流动相的流速等有关。
高效液相色谱的术语

高效液相色谱的术语
高效液相色谱法(High Performance Liquid Chromatography,HPLC)中,有一些常用的术语和概念:
1. 色谱图(Chromatogram):色谱柱流出物通过检测器时所产生的响应信号对时间的曲线图,其纵坐标为信号强度,横坐标为时间。
2. 色谱峰(Peak):色谱柱流出组分通过检测器时产生的响应信号。
3. 峰底(Peak Base):峰的起点与终点之间连接的直线。
4. 峰高(Peak Height):峰最大值到峰底的距离。
5. 峰面积(Peak Area):峰与峰底之间的面积,又称响应值。
6. 基线(Baseline):在正常操作条件下,仅由流动相所产生的响应信号。
7. 基线飘移(Baseline Drift):基线随时间定向的缓慢变化。
8. 基线噪声(Baseline Nois e):由各种因素所引起的基线波动。
以上信息仅供参考,如有需要,建议查阅高效液相色谱相关文献或咨询专业人士。
色谱图和峰参数

色谱图和峰参数色谱图和峰参数Ø色谱图(chromatogram)——样品流经色谱柱和检测器,所得到的信号-时间曲线,又称色谱流出曲线(elution profile)。
Ø基线(base line)——经流动相冲洗,柱与流动相达到平衡后,检测器测出一段时间的流出曲线。
一般应平行于时间轴。
Ø噪音(noise)——基线信号的波动。
通常因电源接触不良或瞬时过载、检测器不稳定、流动相含有气泡或色谱柱被污染所致。
Ø漂移(drift)——基线随时间的缓缓变化。
主要由于操作条件如电压、温度、流动相及流量的不稳定所引起,柱内的污染物或固定相不断被洗脱下来也会产生漂移。
Ø色谱峰(peak)——组分流经检测器时响应的连续信号产生的曲线。
流出曲线上的突起部分。
正常色谱峰近似于对称形正态分布曲线(高斯Gauss曲线)。
不对称色谱峰有两种:前延峰(leading peak)和拖尾峰(tailing peak)。
前者少见。
Ø拖尾因子(tailing factor,T)——T=,用以衡量色谱峰的对称性。
也称为对称因子(symmetry factor)或不对称因子(asymmetry factor)。
《中国药典》规定T应为0.95~1.05。
T<0.95为前延峰,T>1.05为拖尾峰。
Ø峰底——基线上峰的起点至终点的距离。
Ø峰高(peak height,h)——峰的最高点至峰底的距离。
Ø峰宽(peak width,W)——峰两侧拐点处所作两条切线与基线的两个交点间的距离。
W=4σØ半峰宽(peak width at half-height,Wh/2)——峰高一半处的峰宽。
Wh/2=2.355σØ标准偏差(standard deviation,σ)——正态分布曲线x=±1时(拐点)的峰宽之半。
正常峰的拐点在峰高的0.607倍处。
《中国药典》薄层色谱的基本要求

周兰 贵州省食品药品检验所中药(民族药)室
2014年5月
内容
薄层色谱技术在中药标准的优势 《中国药典》薄层色谱的基本要求 薄层色谱应用的影响因素
薄层色谱技术在中药标准的优势
薄层色谱法(《中国药典》2010年版一部 附录Ⅵ B),系将供试品溶液点于薄层板上, 在展开容器内用展开剂展开,使供试品所 含成分分离,将所得色谱图与对照物按同 法所得的色谱图比较,并可用薄层扫描仪 进行扫描,用于鉴别、检查或含量测定的 方法。
饱和方式
方法A(展开缸、薄层板均不饱和) 方法B(展开缸饱和、薄层板不饱和) 方法C(展开缸、薄层板均饱和)
展开-展开剂优化
展开剂在色谱展开中起着关键作用,至今 植物药分析的薄层展开剂的优化基本上还 是依靠经验。主要是因为植物药含有太多 的未知成分,色谱行为各不相同,所以试 错法仍然是中药分析实验室在选择展开剂 时最常用的方法(包括借鉴他人的经验)
(2)分段提取法; 被检测样品不同极性部位分别提取后制成相应供试液, 分别检测分析(如丹参)
(3)特殊提取方式 如蜜丸等含糖较高又有黏性的样品,有机溶剂较难渗透
到样品的内部,可加硅藻土、滑石粉等分散剂共同研匀后 再提取。
供试品溶液净化的方法
(4)液-液萃取法 利用样品与杂质在两相(水相和有机相)中溶解度的不 同,达到净化目的。 特别适合合剂、口服液、糖浆剂、注射剂等的处理。
溶剂总量应足以使薄层板的浸入深度约为5mm(展开剂 量的多少会影响斑点的Rf值)
展开剂要求新鲜配制,一般不用多次反复使用,
如展开溶剂混合放置后有分层现象,则按要求放置分层后 取需要的一相(上层或下层),备用。
展开-饱和与预平衡
色谱图概述——精选推荐

第一章色谱图概述第一节色谱图的获得色谱法(!"#$%&’$(#&)"*)是一种目前使用最广泛的和有效的分离、分析方法。
色谱(!"#$%&’$(#&%)这一概念最早是由俄国植物学家+,-.’’提出,/012年他在一根细长的玻璃管中装入碳酸钙粉末,然后把植物绿叶的石油醚的萃取液倒入管中的碳酸钙上,萃取液的色素就被吸附在管上部的碳酸钙上,再用纯净的石油醚洗脱这些被吸附的色素,于是在碳酸钙上形成了一圈一圈的色带(图34/4/),这些色带被称为色谱,这就是世界上得到的最早的色谱图。
柱中的碳酸钙被称为固定相(,’&’5$6&#*)"&,.),而用作洗脱液的石油醚则被称为流动相(%$758.)"&,.),流动相可以是气体、液体和超临界流体。
将欲被分离的混合物放在固定相上,然后用流动相去洗脱放在固定相上的混合物,这一过程称为洗脱(.89’5$6)。
在洗脱过程中混合物中的不同组分得到分离,陆续从固定相中洗脱下来。
记录这一分离和洗脱过程的图谱就是色谱图34/4/+,-.’’色谱分离示意图(&)刚刚加入萃取液;(7)已加入部分溶剂淋洗图(!"#$%&’$(#&%)。
色谱方法种类很多,分类的方法也很多。
根据流动相是气体还是液体,色谱可分为气相色谱((&,!"#$%&’$(#&)"*)和液相色谱(85:95;!"#$%&’$(#&)"*)。
当流动相是在接近它的临界温度和压力下工作的液体,而这种液体在常温和常压下是气体时,这种流动相就不能简单地说是气体或液体了,这种色谱就称为超临界流体色谱(,9).#!#5’5!&8<895;!"#$%&’$(#&)"*)。
HPLC指纹图谱建立的技术要求

2、共有指纹峰的标定
采用色谱方法制定指纹图谱,必须根据参照物的保留时间,计 算指纹的相对保留时间。根据10批次的检测结果,标定中药材的共有 指纹峰。
3、共有指纹峰面积的比值
(1)以参照物峰面积作为1 (要求能出现稳定的峰) ,计算各共有指 纹峰面积与参照物峰面积的比值(参照物可以是对照品也可以是内标 物)。 (2)以内标物作为参照物的指纹图谱,则以共有指纹峰中其中一个峰 (要求面积相对较大、较为稳定的共有峰)的峰面积作为1,计算其 他各共有指纹峰面积的比值。 (3)各共有峰的面积比值必须相对固定。 (4)共有峰面积要求:单峰面积占共有峰总面积20%的共有峰,其差 值不得20%;若20%面积10%共有峰,其差值不得25%; 若 面积10%的共有峰,峰面积比值不作要求,但必须标定相对保留时 间。未达到基线分离的共有峰,应计算该组峰的总峰面积作为峰面积, 同时标定该组各峰的相对保留时间。
(5)参照物
复方制剂首选君药的活性成分或指标性成分,尽可能阐明参照
物的化学结构及化学名称,也可以选择适当的内标物插入色谱中。
(6)量化的含义
是指在定量的操作条件下,所得到的药材指纹图谱在整体特征 上可以半定量的比较,以表达原料药材指纹图谱在“量”方面的总体 差异程度,以及半成品(提取物)和成品之间及成品批间指纹图谱在纹图谱计算机辅助相似度评价软件》,一 般成品指纹图谱相似度计算结果在0.9-1.0之间为符合要求。
图1 福建莲子HPLC指纹图谱
图2 福建太子参HPLC指纹图谱
第五节 研究实例
丹参HPLC指纹图谱构建 一、药材来源(GAP基地)与鉴定 二、色谱条件与系统适应性试验 用十八烷基硅烷键合硅胶为填充剂;甲醇-0.1%乙酸 (5:95)为流动相;检测波长254nm;柱温为室温;流
色谱简单流程方框图

色谱简单流程方框图:1..典型流程中的各部件2.色谱柱分离原理:利用不同组分在两相间具有不同的分配系数(或吸附平衡常数),当两相作相对运动时,试样中各组分就在两相间反复多次的溶解—解析(或吸附—脱附),使得原来分配系统(或吸附平衡常数)只有微小差别的各组分,产生很大的分离效果,从而将各组分分离开来。
3.色谱操作条件选择最佳流速的选择:从速率理论方程式知道,载气流速对柱效有明显的影响。
如果从小到大改变载气线速,那么它和理论板高H的关系如图(1)所示:μμ图(1)板高H与载气线速μ关系图曲线的最低点,即H最小则柱效最高,此点对应的流速即是最佳线速度。
对N2来说,最佳线速为420~600cm/min;而H2则为600~720cm/min。
在实际工作中,往往采用稍高于最佳线速的流速,以缩短分析时间。
对于一个内径为4mm 的填充柱,载气流速多选用50~80ml/min。
4.固定相的使用温度X围任何一种固定相,都有其使用温度X围。
如柱温超过其上限,则固定相会流失或分解,使柱寿命缩短甚至失效,而且污染检测器;如果低于其下限,则固定液粘度变大,使组分在液相中的扩散系数D L加大,而使传质阻力增高,柱效降低。
往往还会出现异常现象,表现为峰形不正常。
如果低于固定液的凝固点时,则其已不是液相了,失去了分配能力。
一般说来,提高柱温,各组分的挥发度都增加,分配系统变小而组分靠拢,溶剂效率降低,不利于分开。
但操作速度快,分析周期短;降低柱温,有利于分离。
但柱温太低,组分蒸气在两相中的扩散传质速率大为减小,分配不能迅速达到平衡,致使峰形变宽、柱效下降,并延长分析时间。
甚至组分蒸气会冷凝下来,使分析不能正常进行。
5.汽化温度对气化温度的要求:应有足够的温度和热容量使被测试样瞬时汽化。
一般高于柱温50℃以上,或比样品中组分的最高沸点高出20~40℃;试样在该温度下,不被分解。
汽化温度不足的危害:峰形变宽、峰不对称,降低柱效及分离度;峰形异常,不能重复。
色谱系统
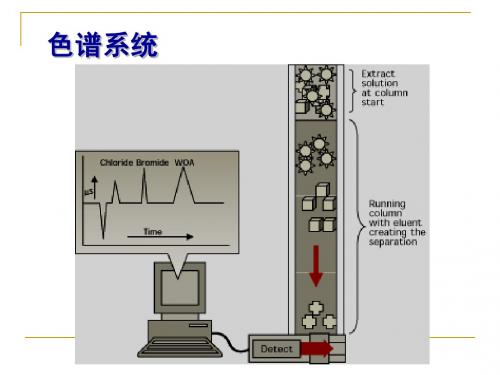
阐明了色谱、蒸馏和萃取之 间的相似性,将色谱柱设想 成由许多液液萃取单元或理 论塔板组成;色谱分离也是 一个分配平衡过程。这就是 Martin等人提出的塔板理论。
(二)塔板理论--衡量柱效率的指标
Martin和Synge 由于对分配色谱的贡献1952年 获得
Nobel 化学奖。
把色谱柱比作一个精馏塔, 沿用精馏塔中塔板的概念来 描述组分在两相间的分配行 为,同时引入理论塔板数作 为衡量柱效率的指标,即色 谱柱是由一系列连续的、相 等水平的塔板组成。每一块 塔板的高度用H表示,称为塔 板高度,简称板高。
当n足够大时,溶质随流动相流过色谱柱。 溶质的移动速度取决于溶质分配在流动相中的 分数。如果溶质较易分配在固定相, 则它的移 动速度较慢。反之,如果溶质较易分配在流动 相,则溶质就容易从色谱柱中流出来。从数学 上说,流动速度取决于q (流动相中的溶质分 数)。 浓度最大的塔板号数rmax,即峰位置:
V0、t0与被测组分无关,因而V’R 、t’R 更合理地反映了物质在柱中的保留情况。
总结:
(1)色谱峰的位置即保留值—进行定
性分析。
(2)色谱峰的h .A —进行定量分析。
(3)色谱峰的位置及峰的宽度—可评 价色谱 柱效的高度。
色谱分析的目的是将样品中各组分彼此分 离,组分要达到完全分离,两峰间的距离必须 足够远。两峰间的距离是由组分在两相间的分 配系数决定的,即与色谱过程的热力学性质有 关。但是两峰间虽有一定距离,如果每个峰都 很宽,以致彼此重叠,还是不能分开。这些峰 的宽或窄是由组分在色谱柱中传质和扩散行为 决定的,即与色谱过程的动力学性质有关。因 此,要从热力学和动力学两方面来研究色谱行
B H A Cu u 该式称为速率方程(范氏方程)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
色谱数据和图谱提交要求
药品注册申报资料所附的色谱数据和图谱的纸面文件可参照国家食品药品监督管理局药品审评中心发布的《药品研究色谱数据工作站及色谱数据管理要求(一)》的相关内容准备,建议对每项申报资料所附图谱前面建立交叉索引表,说明图谱编号、申报资料中所在页码、图谱的试验内容。
用于准备药品注册申报资料的色谱数据的纸面文件应采用色谱数据工作站自动形成的输出文件形式,内容应包括如下相关信息:
一、标明使用的色谱数据工作站,并保留色谱数据工作站固有的色谱图谱头信息,包括:实验者、试验内容、进样时间、运行时间等,进样时间(指injection time)精确到秒,对于软件本身使用“acquired time”、“作样时间”、“试验时间”等含糊表述的,需说明是否就是进样时间。
二、应带有存盘路径的数据文件名。
这是原始性、追溯性的关键信息,文件夹和文件名的命名应合理、规范和便于图谱的整理查阅。
三、色谱峰参数应有保留时间(保留到小数点后三位)、峰高、峰面积、定量结果、积分标记线、理论板数等。
申报资料的色谱数据的纸面文件还应包括色谱数据的审计追踪信息(如色谱数据的修改删除记录及原因)。