高通量测序的生物信息学分析
高通量测序技术的生物信息学分析

高通量测序技术的生物信息学分析引言:高通量测序技术作为一种新型基因测序技术,已经被广泛应用于现代生物研究中。
利用高通量测序技术,科学家们可以快速解码基因组序列、转录组序列以及蛋白质组序列。
然而,高通量测序技术不仅仅是一种实验技术,它也需要强大的生物信息学工具来支持数据分析和解读。
本文将介绍高通量测序技术的生物信息学分析,包括原始数据质量控制、序列比对和注释、基因表达分析、基因变异分析等等。
一、原始数据质量控制原始数据质量控制是高通量测序数据分析的第一步,其目的是剔除低质量序列以及包含污染序列的读段,确保下游的数据分析结果准确可靠。
利用FASTQC等软件对原始的FASTQ格式数据进行质量评估,可以得到关于以下几个质量指标的信息:1. GC含量2. Q20和Q30的比例3. 碱基分布的均匀性4. 过度重叠序列的比例基于以上质量指标,可以对数据进行质量控制处理,包括碱基修剪、低质量序列过滤、去除污染序列等等。
二、序列比对和注释序列比对指的是将高通量测序数据基因组参考序列进行比对,得到参考序列上的SNP、InDel的信息,从而对样品进行全面的基因变异检测、注释和分析。
在序列比对和注释的过程中,需要正确选择合适的比对软件和参考序列。
目前较为流行的比对软件包括BWA、Bowtie、STAR等等。
针对RNA-seq数据的注释工具包括Cufflinks、StringTie、Transcriptome Assembly等等。
基于参考序列的比对结果,还可以利用Variant Effect Predictor (VEP)等工具对候选变异位点进行注释。
注释信息包括dbSNP、ClinVar、ExAC等公共数据库的信息,帮助生物学家了解该变异的生物学特性,并识别其潜在的影响。
三、基因表达分析高通量测序技术还可以用于RNA表达谱的分析,以揭示不同组织和不同发育阶段的基因表达差异。
在基因表达分析中,首先将RNA-seq数据进行质量控制和过滤,然后对序列进行比对和注释,获得基因的计数信息。
生物信息学数据分析的处理流程与方法指南

生物信息学数据分析的处理流程与方法指南概述:生物信息学是一门综合性学科,主要研究生物学信息的获取、存储、处理与分析。
随着高通量测序技术的快速发展,生物信息学数据分析成为了生命科学研究中不可或缺的一个环节。
本文将介绍生物信息学数据分析的处理流程与方法,以帮助研究人员系统地进行生物信息学数据分析。
一、数据预处理生物信息学数据分析的第一步是对原始数据进行预处理。
1. 数据质量控制:对测序数据进行质量控制,去除低质量的碱基和序列,以保证后续分析的准确性。
2. 序列比对:将测序数据与参考基因组或转录组进行比对,确定每个序列的起源以及位置。
二、数据分析数据预处理完成后,可以进行下一步的数据分析,包括以下几个方面:1. 基因表达分析:将转录组数据根据不同条件(如不同时间点、不同处理)进行比较,寻找差异表达的基因。
2. 差异分析:通过比较不同条件下的生物样品,确定差异表达的基因或突变位点。
3. 功能注释:利用公共数据库,对差异表达的基因进行功能注释,寻找其功能以及相关的通路和生物过程。
4. 基因调控网络分析:构建基因调控网络,探究基因之间的关系及其调控网络的重要成员。
5. 蛋白质互作分析:通过蛋白质互作网络,研究蛋白质之间的相互作用,揭示蛋白质的功能及其参与的信号通路。
6. 基因组结构变异分析:研究基因组结构变异,如插入、缺失、倒位等,探究其对个体表型的影响。
7. 代谢组和蛋白组分析:通过代谢组和蛋白组的分析,了解代谢通路和相关蛋白的变化,研究其与生物表型之间的关系。
三、统计分析生物信息学数据分析不可避免地涉及统计分析,帮助我们从数据中找到有意义的关联性或差异。
1. 差异分析的统计学方法:使用适当的统计学方法,如T检验、方差分析等,对差异表达的基因进行统计分析。
2. 多重校正:由于高通量测序数据的量庞大,需要进行多重校正,控制假阳性率。
3. 数据可视化:通过图表或可视化工具,将分析结果直观地呈现,便于研究者理解和解释数据。
生物信息学中的高通量测序数据分析研究

生物信息学中的高通量测序数据分析研究在生物学领域中,随着人们对生物系统的研究日益深入,高通量测序技术的出现为基因和蛋白质序列的鉴定和分析提供了非常有效的手段。
高通量测序数据分析研究作为生物信息学领域中的一个重要分支,以其高效、快速、准确的特性,不断地吸引着研究者的关注。
高通量测序技术是指一种能够同时测定大量DNA序列的方法,它不仅能够用于基因组组装和注释,还可以通过RNA测序技术来分析基因表达。
这种技术极大地加快了生物学研究的速度和效率,为生物学家提供了大量的测序数据。
然而,这些数据的处理和分析却是非常复杂的。
首先,高通量测序数据的预处理是数据分析的第一步。
当测序数据被生成后,必须对其进行质量控制、过滤低质量序列、去除接头序列等处理,才能得到较为准确的数据。
此外,还有一些其他的预处理过程,例如去重、去序列污染等。
接着,对于各类高通量测序数据进行生物信息学分析非常关键。
生物信息学分析的主要目的是确定基因或蛋白质序列,并了解它们在不同生理状态下的表达和功能。
对于RNA测序数据,其主要方法是将原始数据经过拼接或比对成转录本,然后对得到的转录本进行表达量分析,从而确定不同表达和差异表达基因的信息。
而对于DNA测序数据,则可通过基因组拼装、变异分析、基因预测和功能注释等方法进行深入研究。
近年来,生物信息学领域发展迅速,高通量测序数据分析也成为了该领域中的一个热门研究方向。
许多学者正在研究开发更加准确、更加高效、更加专业的分析工具和方法。
例如,结合机器学习技术的表达量分析方法和差异表达基因分析,能够更加准确地发现差异表达的基因;metagenomics(环境微生物组学)领域,则还需要研究多样性分析、代谢通路分析等更为复杂的问题。
总之,高通量测序数据分析是一个关键的研究领域,它为更深入、全面的生物学研究提供了非常重要的工具和方法。
未来,生物信息学领域需要进一步发展,带来更多的高精度、高效率的数据处理和分析技术,以满足更严格的生物学研究需求。
基于高通量测序技术的生物信息学解读

基于高通量测序技术的生物信息学解读高通量测序技术是近年来生命科学和医学领域的重要技术之一,通过对生物样品进行深度测序,能够快速、准确地获取大量生物信息,为基因功能研究、药物研发、疾病诊断和治疗等方面提供了有力支撑。
生物信息学解读是高通量测序技术的重要应用方向之一,涉及到基因组、转录组、蛋白质组等多个层次的分析,具有广泛的研究意义和应用前景。
一、基因组测序基因组测序是高通量测序技术的首要应用方向之一,它能够帮助我们了解生物基因组的组成、结构和功能,为基因功能研究、进化分析等提供数据支持。
与传统方法相比,基因组测序能够在较短时间内对生物基因组进行全面测序,解决了单个基因或单个基因片段测序的限制性问题,提供了更加全面的基因数据。
基因组测序分为宏基因组和微基因组两种。
宏基因组是对各种微生物和大量环境中存在的微生物进行的基因组测序,其目的是揭示微生物种类、多样性、功能等。
微基因组则是对个体或种群的基因组进行的测序,不少研究工作集中在人类基因组的测序上。
基因组测序需要进行序列的拼接、比对、注释等信息学处理才能明确基因组结构和组成。
二、转录组测序转录组是指在细胞内基因转录生成mRNA的总体酶同一时刻产生的所有mRNA分子的总和。
与基因组测序相比,转录组测序可以更加全面地了解生物转录水平的变化。
通过测定生物转录组,可以揭示生物发育、生长、适应环境变化等方面的生物学规律。
在转录组测序中,从样品中提取RNA,然后通过转录组测序技术,对RNA进行深度测序,将结果转化为数字信号,然后进行数据分析和注释,包括基因差异表达分析、聚类分析、功能富集分析等,挖掘生物转录组的生物学意义和作用。
三、甲基化测序在生物体中,DNA甲基化是表观遗传学研究中一个重要的表征,也是人类疾病诊断和治疗的关键因素。
然而,甲基化在不同细胞、组织和环境条件下是动态变化的。
因此,甲基化测序技术可以用来研究DNA甲基化的变化以及与这些变化相关的生物学过程,如基因表达和细胞分化等。
高通量基因测序数据分析及生物信息学算法评估

高通量基因测序数据分析及生物信息学算法评估近年来,随着测序技术的发展和普及,高通量基因测序已成为研究生物学和医学领域的重要工具。
高通量基因测序技术能够迅速、准确地获得大量的DNA或RNA序列数据,为研究人员提供了大量的数据资源,但同时也带来了数据分析和算法评估的挑战。
高通量基因测序数据分析是对产生的海量序列进行处理和解析的过程,其目的是从混合的DNA或RNA样本中准确地识别和描述基因组中的序列变异和表达变化。
这一过程通常包括质量控制、序列比对、变异检测、基因表达分析等步骤。
质量控制是高通量基因测序数据分析的起点,它主要用于检测和过滤掉低质量的序列数据。
低质量的序列数据可能由于测序仪器或实验操作等原因导致错误或偏倚,因此需要通过质量控制筛选出高质量的数据,以保证后续的分析结果准确可靠。
序列比对是高通量基因测序数据分析中的核心步骤之一,它将测序得到的短序列与已知的参考基因组或转录组序列进行比对。
目前常用的比对算法包括BWA、Bowtie等。
比对的目的是确定每个测序片段与参考序列的匹配位置,从而获得关于基因组中变异和表达的信息。
变异检测是高通量基因测序数据分析的重要任务之一。
通过比对结果,可以识别出基因组中的SNP(单核苷酸多态性)、Indel(插入缺失)等变异。
变异检测的方法包括基于规则的变异挖掘、基于统计学的变异检验等,这些方法能够帮助研究人员分析和理解基因组的个体差异。
基因表达分析是高通量基因测序数据分析的另一个重要任务。
通过比对并计算每个基因的表达水平,可以研究基因的功能和调控机制。
常用的基因表达分析方法包括RPKM(Reads Per Kilobase of transcript per Million mapped reads)、DESeq2(Differential Expression analysis based on the Negative Binomial Distribution)等。
高通量测序的生物信息学分析
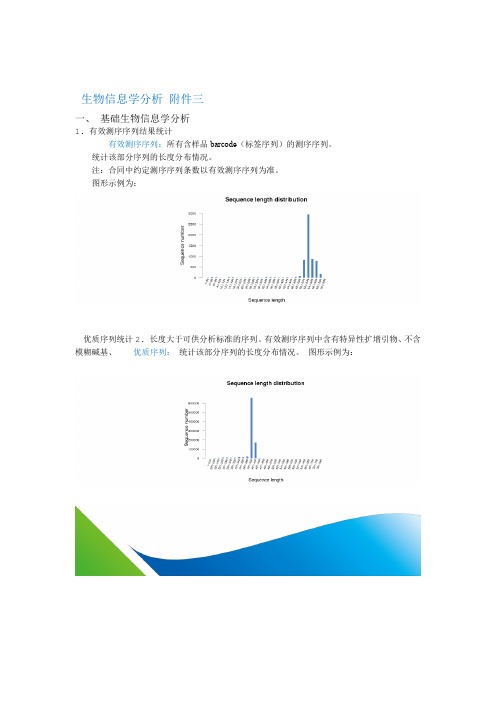
生物信息学分析附件三一、基础生物信息学分析1.有效测序序列结果统计有效测序序列:所有含样品barcode(标签序列)的测序序列。
统计该部分序列的长度分布情况。
注:合同中约定测序序列条数以有效测序序列为准。
图形示例为:优质序列统计2.长度大于可供分析标准的序列。
有效测序序列中含有特异性扩增引物、不含模糊碱基、优质序列:统计该部分序列的长度分布情况。
图形示例为:23.各样本序列数目统计:统计各个样本所含有效测序序列和优质序列数目。
结果示例为:有效序列优质序列样品 AB生成:4.OTU根据序列的相似性,将序列归为多个OTU(操作分类单元),以便后续分析。
HAOTU name EDCBGF101 27 149 OTU1 410 252 45 136 1240 0 0 OTU2 0 0 0 0 029 5 23 1 OTU3 2 17 3 147 11 5 0 OTU4 0 0 47 19 57 45 28 82 OTU5 9 19 3030 0 0 0 0 OTU6 0 0 060 182 5 14 94 24 OTU7 12 00 0 0 0 0 OTU8 0 0 0……………………......……………………5.稀释曲线(rarefaction 分析)根据第4条中获得的OTU数据,做出每个样品的Rarefaction曲线。
本合同默认生成OTU 相似水平为0.03的rarefaction曲线。
rarefaction曲线结果示例:3指数分析6.计算各个样品的相关分析指数,包括:ace\chao丰度指数:?shannon\simpson多样性指数:? 0.03的上述指数值。
本合同默认生成OTU?相似水平为多样性指数分析结果示例:注:默认分析以上所列指数,如有特殊需要请说明。
Shannon-Wiener曲线7.利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线,反映各样本在不同测序数量时的微生物多样性。
高通量测序生物信息学分析(内部极品资料,初学者必看)

基因组测序基础知识㈠De Novo测序也叫从头测序,是首次对一个物种的基因组进行测序,用生物信息学的分析方法对测序所得序列进行组装,从而获得该物种的基因组序列图谱。
目前国际上通用的基因组De Novo测序方法有三种:1. 用Illumina Solexa GA IIx 测序仪直接测序;2. 用Roche GS FLX Titanium直接完成全基因组测序;3. 用ABI 3730 或Roche GS FLX Titanium测序,搭建骨架,再用Illumina Solexa GA IIx进行深度测序,完成基因组拼接。
采用De Novo测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及进行比较基因组学研究,为后续的相关研究奠定基础。
实验流程:公司服务内容1.基本服务:DNA样品检测;测序文库构建;高通量测序;数据基本分析(Base calling,去接头,去污染);序列组装达到精细图标准2.定制服务:基因组注释及功能注释;比较基因组及分子进化分析,数据库搭建;基因组信息展示平台搭建1.基因组De Novo测序对DNA样品有什么要求?(1) 对于细菌真菌,样品来源一定要单一菌落无污染,否则会严重影响测序结果的质量。
基因组完整无降解(23 kb以上), OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;每次样品制备需要10 μg样品,如果需要多次制备样品,则需要样品总量=制备样品次数*10 μg。
(2) 对于植物,样品来源要求是黑暗无菌条件下培养的黄化苗或组培样品,最好为纯合或单倍体。
基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。
(3) 对于动物,样品来源应选用肌肉,血等脂肪含量少的部位,同一个体取样,最好为纯合。
基于高通量测序技术的37种HPV亚型流行特征鉴定与生物信息学分析

基于高通量测序技术的37种HPV亚型流行特征鉴定与生物信息学分析高通量测序技术(high-throughput sequencing)是一种快速、准确、高效的基因组学研究方法,被广泛应用于各个领域,包括人类疾病的分子机制研究。
本文将基于高通量测序技术,对37种人类乳头状瘤病毒(HPV)亚型的流行特征进行鉴定,并进行相应的生物信息学分析。
首先,我们需要明确37种HPV亚型的定义和分类。
HPV是一类双链DNA病毒,被广泛认为是导致宫颈癌和其他一些肿瘤的主要原因之一。
根据其基因组序列的相似性,HPV亚型被分为多个不同的类型。
具体的亚型包括HPV16、HPV18、HPV31、HPV33等,共计37种。
接下来,我们将使用高通量测序技术对37种HPV亚型进行测序。
高通量测序技术的优势在于可以高效地测定一次性产生的大量DNA序列,从而揭示基因组的整体特征。
通过测序,我们将获得每种亚型的基因组序列信息,并对其进行初步的分析。
在测序完成后,我们将对这些基因组序列进行生物信息学分析,以鉴定HPV亚型的流行特征。
生物信息学是一门将计算机科学和统计学方法应用于生物学研究的交叉学科,借助它的帮助,我们可以挖掘和分析大规模基因组数据中的有用信息。
在分析过程中,我们可以利用生物信息学工具对这些HPV亚型的基因组序列进行比对和序列标识。
通过比对,我们可以发现亚型之间的相似性和差异性,从而揭示其流行特征。
此外,还可以使用聚类分析方法将HPV亚型分成不同的群组,推断其可能的发生传播路径。
另外,我们还可以进行亚型基因组中功能区域的注释和预测。
通过分析基因组序列中的开放阅读框(open reading frame,ORF),我们可以预测编码蛋白质的基因,并进一步注释这些编码蛋白质的功能。
此外,还可以预测亚型的结构域和功能位点,为后续研究提供有价值的信息。
最后,我们可以利用系统生物学方法研究HPV亚型的调控网络和相互作用网络。
系统生物学是一种研究生物系统中各个组成部分之间相互作用与调控的学科,它可以揭示基因之间的调控关系和信号传导路径。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附件三生物信息学分析
一、基础生物信息学分析
1.有效测序序列结果统计
有效测序序列:所有含样品barcode(标签序列)的测序序列。
统计该部分序列的长度分布情况。
注:合同中约定测序序列条数以有效测序序列为准。
图形示例为:
2.优质序列统计
优质序列:有效测序序列中含有特异性扩增引物、不含模糊碱基、长度大于可供分析标准的序列。
统计该部分序列的长度分布情况。
图形示例为:
3.各样本序列数目统计:
统计各个样本所含有效测序序列和优质序列数目。
结果示例为:
样品有效序列优质序列
A
B
4.OTU生成:
根据序列的相似性,将序列归为多个OTU(操作分类单元),以便后续分析。
OTU name A B C D E F G H
OTU1 149 410 27 252 45 124 136 101
OTU2 0 0 0 0 0 0 0 0
OTU3 2 3 14 23 1 5 17 29
OTU4 0 47 0 11 0 5 1 7
OTU5 19 28 82 9 57 45 303 9
OTU6 0 0 0 0 0 0 0 0
OTU7 0 182 94 24 14 5 12 60
OTU8 0 0 0 0 0 0 0 0
...... …………………………………………
5.稀释曲线(rarefaction 分析)
根据第4条中获得的OTU数据,做出每个样品的Rarefaction曲线。
本合同默认生成OTU相似水平为0.03的rarefaction曲线。
rarefaction曲线结果示例:
6.指数分析
计算各个样品的相关分析指数,包括:
•丰度指数:ace\chao
•多样性指数:shannon\simpson
•本合同默认生成OTU相似水平为0.03的上述指数值。
多样性指数分析结果示例:
注:默认分析以上所列指数,如有特殊需要请说明。
7.Shannon-Wiener曲线
利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线,反映各样本在不同测序数量时的微生物多样性。
当曲线趋向平坦时,说明测序数据量足够大,可以反映样品中绝大多数的微生物信息。
绘制默认水平为:0.03。
例图:
8.Rank_Abuance 曲线
根据各样品的OTU丰度大小排序作丰度分布曲线图。
结果文件默认为PDF格式(其它格式请注明)。
例图:
9.Specaccum物种累积曲线(大于10个样品)
物种累积曲线( species accumulation curves) 用于描述随着抽样量的加大物种增加的状况,是理解调查样地物种组成和预测物种丰富度的有效工具,在生物多样性和群落调查中,被广泛用于抽样量充分性的判断以及物种丰富度( species richness) 的估计。
因此,通过物种累积曲线不仅可以判断抽样量是否
充分,在抽样量充分的前提下,运用物种累积曲线还可以对物种丰富度进行预测。
10.样品OTU分布及分类学信息
OTU产生后,统计各个样品含有OTU情况及每个OTU中含有序列的数目。
同时,将所有序列与Silva库比对,得到序列的分类学信息。
通过寻找最近祖先方法,得到每个OTU的分类学信息。
本合同默认分析相似性水平为0.03的OTU。
结果为一份xls文件,文件内容示例为:
第一列为OTU编号,第一行为各个样品名称,中间数字表示该列样品在此行OTU中所占的序列数目,最后一列为该行OTU的种属信息。
二、高级生物信息学分析
11.OTU 分布VENN图
注:选择一组不多于五个样品,分析样品间OTU重合情况,将结果以VENN图形式展示。
结果
文件默认为PDF格式(其它格式请注明)。
例图:
12.多样品相似度树状图I (样品无分组)
注:选定需要分析的多个样品作为一组对比分析,使用jest算法,比较该组分析中各样品在OTU
(0.03)水平上的群落结构相似度并作出树状图。
结果文件默认为PDF格式(其它格式请注明)。
例图:
13.多样品相似度树状图II (样品有分组)
注:选定需要分析的多个样品作为一组对比分析,使用jest算法,比较该组分析中各样品在OTU
(0.03)水平上的群落结构相似度并作出树状图。
结果文件默认为PDF格式(其它格式请注明)。
例图:
14.群落结构组组分图(共__N__组分析)
注:选定一个或多个需要分析的样品,选定一个分类学水平,按照相应多样性信息作图,反应各样品的群落结构。
结果文件默认为PDF格式(其它格式请注明)。
可选分类学水平:门、纲、目、科、属;同一组样品选择多个分类学水平为多组分析。
例图:
15.多样品相似度树与柱状图组合分析(默认提供门的水平)
左边是样品间基于群落组成的层次聚类分析,右边是样品的群落结构柱状图。
16.PCA主成分分析Ⅰ(样品无分组)
选取多个样品,进行PCA分析。
结果文件默认为PDF格式(其它格式请注明)。
例图:
17.PCA主成分分析Ⅱ(样品有分组信息)
选取多个样品,进行PCA分析。
结果文件默认为PDF格式(其它格式请注明)。
例图:
18.群落Heatmap图(确定分类学水平及图片颜色)
选择多个样品,作出其在选定的分类学水平上群落结构Heatmap图。
结果文件默认为PDF格式(其它格式请注明)。
图形颜色默认为彩虹色,如需选黑红色请标明。
可选分类学水平:门、纲、目、科、属、OTU(0.03)
如分析单元数目较多,默认使用序列数较多的前100个种属或OTU作图,如有其它要求请注明。
例图:。