DNA测序结果拼接方法
DNAman8序列比对、序列拼接图文教程
DNAman8序 列 比 对 、 序 列 拼 接 图 文 教 程
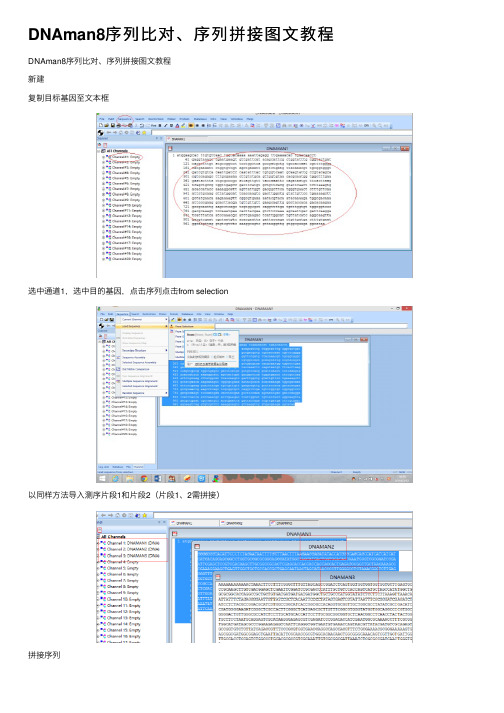
DNAman8序列比对、序列拼接图文教程 新建 复制目标基因至文本框
选中通道1,选中目的基因,点击序列点击from selection
以同样方法导入测序片段1和片段2(片段1、2需拼接)
拼接序列
点击通道 勾选需要拼接序列
点击拼接,点击显示结果
点击export输出拼接后序列 选中并参照上述方法拼接序列导入通道序列比对 多重序列比对
选中目标基因和拼接后基因通道 一直下一步下一步完成
如果测序样品很多,可以依次导入进行多重对比,点击options可以选择对比对象
3730测序结果说明

测序结果说明1.测序完成后,我们用对每个样品提供一份测序报告,其中包括:测出的序列彩色峰图(请用Chromas软件打开)序列文件拼接后结果(需要测通的样品)2.在进行DNA测序时,紧接引物的10~30碱基有时不一定能完全读清楚。
3.正常情况下,3730测序仪保证800bp的有效长度,但是有时由于DNA结构上的原因,有时会出现反应中断无法进行的情况。
如:G/C rich;G/C Cluster;Poly A/T/C/G的连续结构等。
此外,另一种情况为反应中途出现的套峰现象,此种情况一般为DNA结构中的重复序列,造成测序用引物和模板之间有两个以上结合位点。
以上情况是由于DNA结构原因造成了无法正常进行DNA测序,对这种情况,我们会根据具体测序结果进行相应收费。
出现以上情况后,我们提倡从另一端进行测序,或者用高级试剂盒进行测序。
4.备注说明一般正常的菌液和质粒自收样日至发送报告日周期为二--三天,PCR产物(纯化及未纯化)为三--四天。
对于三个工作日内无法得到满意测序结果的,我们会用E-mail或电话与客户或代理商联系。
并不是所有的样品都能得到满意的测序结果,对于测序结果不好的,我们会尽力找到可能的原因,以建议进一步如何操作。
测序常见问题解答Q1.为什么提供新鲜的菌液?如何提供新鲜的菌液?A1.首先,新鲜的菌液易于培养,可以获得更多的DNA,同时最大限度地保证菌种的纯度。
如果您提供新鲜菌液,用封口膜封口以免泄露;也可以将培养好的4~5ml菌液离心沉淀下来,倒去上清液以方便邮寄。
同时邮寄时最好用盒子以免邮寄过程中造成离心管挤压破裂。
Q2.DNA测序样品用什么溶液溶解比较好?A2.溶解DNA测序样品时,用灭菌蒸馏水溶解最好。
DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件。
如果DNA用缓冲液溶解后,在进行测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。
染色体水平组装基因组

染色体水平组装基因组染色体水平组装基因组是一种重要的生物学技术,它可以帮助我们更好地理解基因组的结构和功能。
本文将介绍染色体水平组装基因组的原理、方法和应用,并探讨其在生物学研究和医学领域的潜在应用。
染色体水平组装基因组是指通过将测序读段按照染色体上的位置进行组装,重建出完整的染色体序列。
相比于传统的基因组组装方法,染色体水平组装基因组能够提供更长的连续序列,有助于揭示基因组的结构和功能。
染色体水平组装基因组的原理是利用测序技术对DNA分子进行测序,并根据测序结果将读段按照染色体上的位置进行组装。
首先,需要将DNA分子进行打断,并利用测序技术对其进行测序。
然后,根据测序结果将读段按照染色体上的位置进行排序和组装。
最后,通过对组装结果进行验证和校正,得到完整的染色体序列。
染色体水平组装基因组的方法主要包括两个步骤:测序和组装。
测序步骤可以采用多种测序技术,如Sanger测序、Illumina测序和PacBio测序等。
不同的测序技术具有不同的优缺点,可以根据研究的需求选择合适的测序技术。
组装步骤则是将测序读段按照染色体上的位置进行排序和组装,常用的组装算法包括Overlap-Layout-Consensus(OLC)算法和De Bruijn图算法等。
染色体水平组装基因组在生物学研究中具有广泛的应用。
首先,它可以帮助我们理解基因组的结构和功能。
通过组装染色体序列,我们可以了解基因的分布和排列方式,揭示基因组的整体结构和组织方式。
其次,染色体水平组装基因组可以帮助我们研究基因组的进化和变异。
通过比较不同物种的染色体序列,我们可以揭示物种间的遗传差异和进化关系。
此外,染色体水平组装基因组还可以应用于基因组编辑和合成生物学等领域,为基因工程和合成生物学的研究提供重要的工具和方法。
在医学领域,染色体水平组装基因组也具有重要的应用价值。
首先,它可以帮助我们研究人类基因组的结构和功能。
通过组装人类染色体序列,我们可以了解人类基因的分布和排列方式,揭示人类基因组的整体结构和组织方式。
简述一、二、三代测序技术

简述一、二、三代测序技术
一代测序技术
一代测序技术是一种拼接式测序技术,它可以将DNA片段进行拼接,从而得到DNA序列。
它是一种基于Sanger方法的技术,通过热板和冷板将DNA片段分别固定在支架上,再使用DNA聚合酶对支架上的DNA片段进行复制,最后通过测序仪来获取DNA序列信息。
一代测序技术已经被广泛应用于基因组学研究中,但是它仍然有很多缺点,比如时间短,费用较高,最大的问题是在测序过程中可能出现错误,这种错误很难被确认。
二代测序技术
二代测序技术是一种新的技术,它不需要DNA片段的拼接,而是使用DNA分子组装的方法来提取DNA序列信息。
该技术使用高通量测序技术,可以一次性同时测序大量的DNA片段,因此大大提高了测序效率,并减少了出错的几率,同时也降低了测序成本。
三代测序技术
三代测序技术是一种后续的测序技术,它能够更加精确地提取DNA序列信息,使用特殊的测序仪可以同时测定全基因组的DNA序列。
该技术采用短片段拼接的方法,可以实现更高精度的DNA序列测序,可以更好地发掘基因组中的变异位点,从而更好地研究遗传病和肿瘤的发生机制。
三代测序拼接算法

三代测序拼接算法(原创版)目录1.三代测序拼接算法的背景和意义2.三代测序拼接算法的原理和方法3.三代测序拼接算法的应用案例4.三代测序拼接算法的优缺点和未来发展方向正文三代测序拼接算法是一种在基因组学研究中广泛应用的技术,尤其在处理较长的 DNA 序列拼接上具有重要意义。
本文将从原理、方法、应用案例以及优缺点等方面,详细介绍三代测序拼接算法。
一、三代测序拼接算法的背景和意义随着基因组学研究的深入,研究人员需要对越来越长的 DNA 序列进行拼接。
传统的 Sanger 测序技术由于其局限性,难以应对这种需求。
因此,三代测序拼接算法应运而生,它能够更有效地处理较长的 DNA 序列拼接问题。
二、三代测序拼接算法的原理和方法三代测序拼接算法主要基于 PacBio SMRT 技术,通过构建 SMRT 测序数据和 Hi-C 数据之间的联系,实现长 DNA 序列的拼接。
具体方法包括以下几个步骤:1.构建 SMRT 测序数据和 Hi-C 数据的联系通过比对 SMRT 测序数据和 Hi-C 数据,找到它们之间的匹配区域,从而构建起它们之间的联系。
2.利用联系进行拼接根据构建的联系,将 SMRT 测序数据和 Hi-C 数据进行拼接,得到目标 DNA 序列。
3.拼接结果评估与优化对拼接结果进行评估,通过优化拼接策略和参数,提高拼接的准确性和完整性。
三、三代测序拼接算法的应用案例三代测序拼接算法在多个领域都取得了显著的应用成果,例如:1.人类基因组拼接利用三代测序拼接算法,研究人员成功拼接了人类基因组中的复杂区域,为全面解析人类基因组结构提供了有力支持。
2.动植物基因组拼接三代测序拼接算法在动植物基因组拼接方面也取得了显著成果,为研究动植物基因组结构和功能提供了有力工具。
四、三代测序拼接算法的优缺点和未来发展方向三代测序拼接算法具有以下优缺点:优点:能够有效地处理较长的 DNA 序列拼接问题,提高拼接的准确性和完整性。
DNA测序结果中常见的几个问题

1 、为什么开始一段序列的信号很杂乱,几乎难以辨别这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生 N 值,峰图较杂由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生 N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)是否会影响测序结果序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
7 、测序结果为什么与标准序列有差别原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。
DNA测序 化学降解法

每个单链的同一方向末端都用放射性同位素标记,以
便显示DNA条带
↓
分别用不同方法处理,获得只差一个核苷酸的降解
DNA群体
↓
电泳,读取DNA的核苷酸顺序
8
2021/6/16
9
① 先用限制性内切酶把DNA切成10—200bp 的测序
材料;
② 用碱性磷酸化酶处理该片段,消除5′末端上的磷酸;
③ 在5′OH端标记 32P,用多核苷酸磷酸激酶催化;
0.03mol/LNaOH、10%甘油、1mmol/L EDTA、
0.05%二甲苯蓝和0.05%溴甲酚蓝的溶液中,
使双链DNA变性。然后加到由5%~10%的丙烯
酰胺、0.16%~0.33%的双丙烯酰胺、
50mmol/LTris-硼酸、pH8.3的1mmol/L EDTA
组成的凝胶板上,进行电泳和放射性自显影等
的核苷酸序列,相互参照测定结果,可以得到准
确的 DNA 链序列。
2021/6/16
33
缺点:
没有改进,操作繁琐, 化学试剂的毒性大,放
射性同位素标记效率偏低以致需要较长的放
射自显影曝光时间,人工读取数据费时费力.
目前,仅在分析特殊DNA链的核苷酸序列和
分析DNA和蛋白质相互作用中的DNA一级结
构时才使用
直接或间接特异性识别4种碱基特定化学试剂可对碱基进行特异性修饰在修饰碱基处5或3打断磷酸二酯键将一个dna片段的端磷酸基作放射性标记再分别采用不同的化学方法修饰和裂解特定碱基从而产生一系列长度不一而端被标记的dna片段这些以特定碱基结尾的片段群通过凝胶电泳分离再经放射线自显影确定各片段末端碱基从而得出目的dna的碱基序列
优点:不需要进行酶催化反应,因此不会产
基因序列拼接算法设计(精)

1 . 2 分析模块
分析模块包括对输入的数据进行预处理如数据中小写字 母统一转换为相应的大写字。然后根据杂交匹配出的探针, 利用字符串的相关操作命令, 拼接重组出靶序列的互补序列, 再对互补序列字符串中的 A与 T 、 G与 C进行互补替换, 得到 靶序列。
收稿日期: 2 0 0 9 - 1 0 - 0 8 。国家自然科学基金( 3 0 6 7 1 8 7 2 , 3 0 7 7 1 8 9 9 ) 。
图3 拼接分支示意图
1 软件主要功能模块与结构
1 . 1 数据输入模块
该模块实现匹配探序列的输入功能, 由于测序结果通常是 以文本文件提交, 因此设计了可读入文本格式数据的功能。 另外, 为了验证软件的分析是否正确, 还设计了随机生成给 定长度 D N A序列的功能, 同时根据生成的 D N A序列和设定的 探针长度, 自动得出匹配探针, 以便后续的拼接处理, 主研领域: 信号与信息处理, 图像处理。
第 5期 1 . 3 拼接的处理过程
刘国庆等: 基因序列拼接算法设计
2 5
分支继续进行拼接。例如在图 3中, 分支点 1处的位置为 1 0 , 用 P U S H函数将数字 1 0压入堆栈, 然后将分支点 1处的 A 、 G字符 排序, 选择字符 A继续进行拼接。到分支点 2处时, 该处位置 为1 5 , 将该数字压入堆栈, 选择字符 C继续进行拼接。 当拼接出来的 D N A链满足一定条件( 此条件将在第 3节中 讨论) , 则输出结果并存入列表框控件中。并且检查堆栈中有 无数据, 如果有, 则弹出堆栈中最上面的数据 ( 即最后压入栈 的) , 得到分支位置。然后根据此位置数据, 进行如下操作: ( 1 ) 从已拼接的 D N A链中获取该位置前的字符串, 以便从 该处开始拼接; ( 2 ) 从已拼接的 D N A链中获取该位置前 4个字符( 即探针 长度减 1 ) ; ( 3 ) 从匹配探针列表中, 查找前 4个字符与第 2步中所得 的字符串相同的匹配探针, 从已拼接的 D N A链中判断该探针是 否已使用, 如果未使用, 则用该探针继续拼接。 重复按上面的步骤, 直到堆栈为空, 拼接结束。 这一算法是将所有匹配探针作为起始探针进行尝试拼接, 计算量明显比较大, 可以考虑在靶 D N A链的 5 ’ 端挂一较短长 度且碱基序列已知的寡核苷酸片段。由于 D N A链的 5 ’ 端是起 始端, 因此该链和基因芯片进行杂交匹配后, 起始匹配探针必定 是所挂的寡核苷酸片段互补序列的前端部分, 从该探针开始拼 接, 可以大幅减少拼接运算的计算量。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
粘贴下载的序列
填写你想拼 接的序
列。。。进 行blast
选择一个序列,进入
将序列下载下来
点击blast
点击
粘贴序列 选择otherS
填写想比对的物种
选择序列
运用Dnaman
序列--序列拼接
线ORF
点击
粘贴拼接好 的序列
点击
点击查 看详情
点击
结果
谢谢观看! 2020
待拼接序列显示区
点击
运用PBIL进行在线拼接
粘贴要拼接的 两个序列
点击
点击Contigs,即可出现 拼接好的序列
运用DNAMAN进行 本地ORF
搜索—开放阅读框 文件--打开--(拼接好的序列)
搜索—开放阅读框--(得到的东西)
蛋白质—翻 译纵览(结
果)
点击(得到的结果)